
Abstract
We present a simple method based on transmission electron microscopy that allows investigation of the early steps of polyplex-mediated transfection without the use of labeled DNA. The ultrastructural analysis showed internalization of 0.2–1-μm aggregates composed of 30–50-nm subunits. In addition, new details of the internalization process were revealed, suggesting an unspecific cell entry mechanism of large DNA aggregates.
T
Peptide-mediated gene transfer was used as a model system to transfect human cancer cells in vitro (Dalluge et al. 2002). The synthetic peptide (K16-NLS; CKKKKKKKKKKKKKKKKGGGPKKKRKVG) contained 16 lysine residues for binding to the DNA, and a C-terminal nuclear localization signal (NLS) derived from the SV 40 T large antigen (Cartier and Reszka 2002) was used to facilitate nuclear transport of the peptide/DNA complex. Complexes (lysine/phosphate ratio 1.5) were formed by mixing 2 μg of plasmid DNA (pLUC, ∼6.5 kb) (Cartier et al. 2000) with 0.43 nmol peptide in 100 μl physiological Tris buffer (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 4 mM CaCl2) and subsequent incubation at room temperature for 30 min.
We examined the peptide/DNA complexes after their formation in solution using transmission electron microscopy. Complexes were stained with uranyl acetate on a formvar-coated grid (Bremer and Rasquin 1998) and analyzed with a Zeiss EM 900 at an accelerating voltage of 80 kV. The complexes were observed as a heterogeneous population of aggregates ranging from 0.3 μm to 3 μm in size and composed of 30–70 nm spherical subcomplexes (Figures 1A–1C). This is in agreement with similar studies using polylysine or polyethyleneimine (Tang and Szoka 1997).
The complexes used in this study have a positive ζ potential (data not shown) and therefore can potentially interact with serum proteins. Hence, we investigated the morphology of the complexes that sedimented onto the cell surface in vitro by scanning electron microscopy. RPMI medium (900 μl) containing 10% FCS, 100 μM chloroquine, and 4 mM CaCl2 was added to the complexes, transferred onto 4 × 105 human colon carcinoma cells HCT 116, and incubated at 37C. Chloroquine was used to facilitate endosomal escape of endocytosed DNA complexes. CaCl2 was added as an enhancing factor of in vitro transfection systems (Haberland et al. 1999). Preliminary transfection experiments using the luciferase reporter gene showed that the transfection efficiency of the present system was comparable to that obtained after lipofectin-mediated transfection (data not shown).
At 30 min after transfection, the cells were fixed with 2% glutaraldehyde (Serva; Heidelberg, Germany) in 0.1 M phosphate buffer for 20 min. Then the samples were washed twice in PBS and dehydrated in a graded series of acetone (25, 50, 70, 90, 100%) before being critical point-dried. The samples were then coated with a 17-nm gold/palladium layer (80 sec, 40 mA) in an SCD 050 sputter coater (Balzers) and examined in a JEOL 25SIII scanning microscope operated at 25 kV. The peptide/DNA complexes observed on the cell surface (Figures 1D and 1E) shared the same size and morphology as those detected in solution (Figure 1A). This indicates that the serum present in the transfection medium does not induce any apparent alterations of the aggregates.
Next we investigated the internalization of the complexes and their intracellular fate at the ultrastructural level. A simple method based on transmission electron microscopy was adapted from Russel and Burguet (1977), allowing direct visualization of the complexes without the need to use chemically labeled DNA. After fixation in 2% glutaraldehyde and post-fixation in 1% OsO4 and 1.5% K4Fe(CN)6, samples were dehydrated in a graded alcohol series and embedded in Epon 812 epoxy resin (Fluka; Buchs, Switzerland). The dehydration steps were performed in the presence of 1% p-phenilendiamine (Roth; Karlsruhe, Germany) in 70% alcohol to achieve maximal lipid preservation. Sections of 70 nm were cut using a Reichert-Jung Ultracut E ultramicrotome (Leica UK; Milton Keynes, UK), collected on copper grids, and stained with uranyl acetate and lead citrate before examination in a Zeiss EM 900 at 60 or 80 kV acceleration voltage.
Thirty minutes after transfection, dark stained aggregates were found close to the cell surface (Figure 2A) or located in plasma membrane invaginations (Figure 2C), suggesting an ongoing internalization. Aggregates were also found inside the intracellular compartments (Figures 2B and 2D). Examination of 100 sections from three different experiments showed that the size of internalized aggregates varied from 0.2 μm to 1 μm. We cannot exclude the possibility that the fixation procedure used in this study induces aggregation of the complexes at the cell surface and also inside the endosomal compartment. Therefore, the actual size of the internalized material may be smaller and may originate from single or oligomeric DNA complexes. However, complexes that are not connected to large aggregates represent only a relatively small portion of the total complex population found in solution. Considering the relatively large amount of the internalized material, it is more likely that large aggregates are actually internalized. Therefore, we hypothesize that under the present conditions the tumor cells behave as non-professional phagocytes able to ingest large particles.
To demonstrate that the observed aggregates were in fact the DNA complexes prepared before transfection, the same experiments were performed using digoxigenin-labeled plasmid DNA (Zaitsev et al. 2002). At 4 hr after transfection, the cells were fixed and processed for immunogold detection of the labeled DNA as described previously (Haberland et al. 1999). Cryosections were contrasted and stabilized either conventionally with 0.3% uranyl acetate and 2% methylcellulose (25 cps; Sigma, Deisenhofen, Germany) according to Tokuyasu (1980,1986) or with a mixture of 3% tungstosilicic acid hydrate (Fluka) and 2.5% polyvinylalcohol (Mr 10,000; Sigma) according to Kargel et al. (1996). Electron micrographs were obtained with a Zeiss EM 910 at an acceleration voltage of 80 kV. Dense peptide/DNA complexes labeled with gold particles were found attached to the cell surface (Figure 2E) and in intracellular compartments (Figure 2F). Sections of control cells without DNA were always free of label (not shown). In addition, the similarity of the structures in terms of size and morphology to the complexes containing unlabeled DNA (Figures 2A–2D) provides indirect evidence that, in the procedure using unlabeled DNA, the internalized material corresponds to the DNA complexes used for transfection.
The positive ζ potential of the complexes also suggests their direct binding to the cell surface and subsequent cell entry via phagocytosis. According to the classical zipper-type phagocytosis model, one may expect extension of pseudopods along the particles, resulting in continuous contact between the particle and the cell membrane or the vacuole wall. However, at higher magnification only a few focal contact points were observed (Figures 3A and 3B), and the internalized material was located in relatively spacious vacuoles shortly after engulfment (Figure 2D). Nevertheless, we cannot exclude a possible attachment to the surrounding glycocalyx, which is not visible using this technique. Additional experiments would be necessary to analyze the role of the glycocalyx during the internalization process. Receptor-mediated cell uptake involves the recruitment of cellular factors at the internalization site of the membrane. However, ultra-structural changes accompanying this process were not observed (Figures 3A and 3B).
The intracellular pathway was further examined using both methods with labeled and unlabeled DNA (Figures 3C–3E). At 4 hr after transfection, large vacuoles containing DNA were observed. The morphology of the DNA-containing structures suggests an ongoing degradation of a large aggregate (Figure 3C). Vacuoles with dark condensed structures were also observed (Figures 3D and 3E). However, no DNA was identified within these structures (Figure 3D), suggesting an advanced degradation stage. This result also shows that the technique based on simple contrast staining is more suitable to study early steps of transfection before degradation of the DNA takes place. Taken together, these observations suggest a cell entry process by a nonspecific phagocytosis-like mechanism that leads to the fusion of phagosomes with the lysosomal compartment.
Chloroquine is used as an enhancing reagent in polyplex-mediated transfection, but its mechanism of action is not completely understood. It is believed to prevent acidification of endosomes or lysosomes, which inhibits the activation of degradative enzymes and reduces the degradation of endocytosed DNA complexes (Erbacher et al. 1996). We examined whether the effect of chloroquine is accompanied by intracellular morphological changes. When chloroquine was omitted from the transfection medium, a significantly larger number of relatively smaller vacuoles was observed in the cytoplasm after 4 hr of incubation (Figure 3F). In the presence of chloroquine, only one or two relative large vacuoles were found located in the perinuclear region (Figures 3C–3E). This observation is in agreement with other studies showing a chloroquine-induced perinuclear accumulation of endosomes containing transfected DNA (Erbacher et al. 1996). Therefore, we propose that the enhancing effect of chloroquine relies on both the protection of the vector from degradation and an altered trafficking of the endosomes facilitating transport to the perinuclear region.
Calcium has been shown to enhance the efficiency of histone- and cationic peptide-based transfection systems, probably by inducing dissociation of the DNA complex in the endosome and also by facilitating its release into the cytoplasm (Zaitsev et al. 2002). In principle, calcium may also influence the morphology of DNA complexes in the transfection medium as well as early steps of the transfection process, including cellular uptake. To detect morphological changes induced by the calcium, control experiments omitting calcium in the transfection medium were performed. No apparent differences in the ultrastructure or the overall amount of internalized material were found. Alterations in the intracellular pathway of the complexes were also not found using our technique (data not shown). This confirms a possible effect of calcium at later stages of the gene transfer, most likely after the DNA escaped into the cytoplasm. Interestingly, in control experiments that included calcium in the transfection medium but without the addition of DNA complexes and chloroquine, an increased number of dying mitochondria located in autophagosomes were observed (Figure 3G), illustrating the cytotoxicity of the calcium. In addition, the degraded mitochondria were identical to the condensed structures found in the vacuoles 4 hr after transfection when DNA complexes and calcium (Figures 3D and 3E) were added. This suggests a fusion between phagosomes containing the internalized complexes and autophagosomes containing damaged mitochondria at later stages of the transfection process.

(
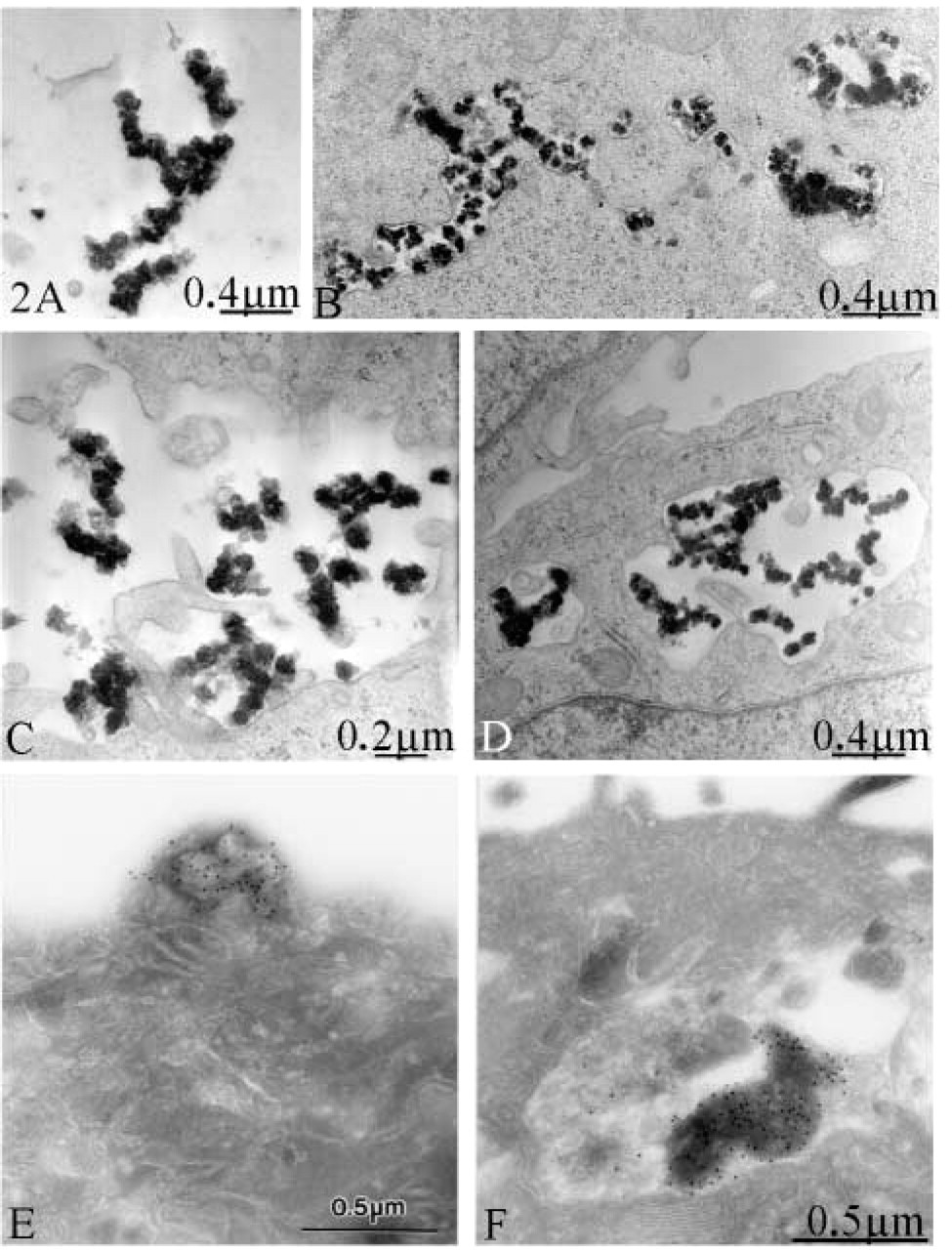
Morphological observations of peptide/DNA complexes during transfection of HCT 116 cells. (
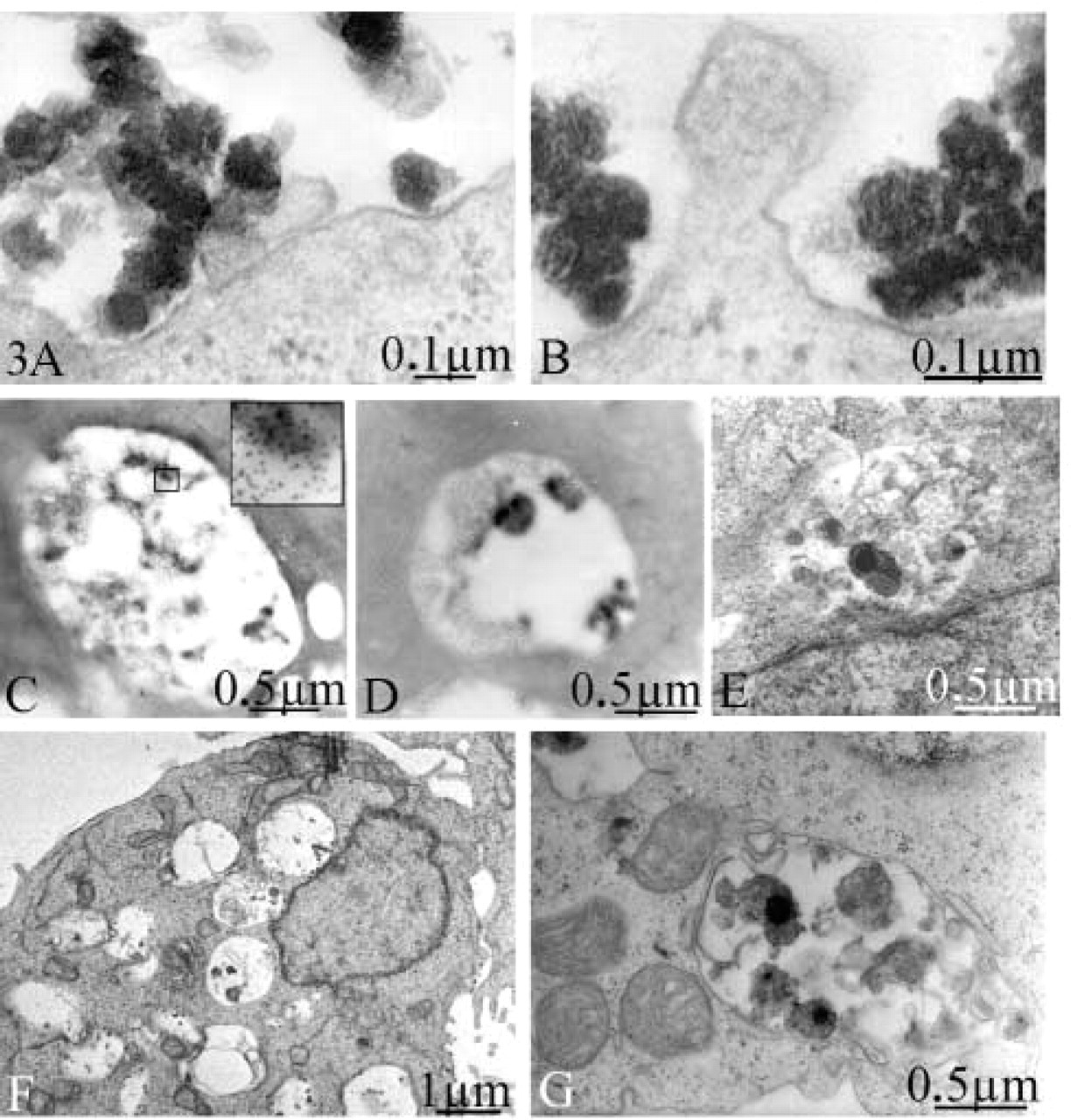
Ultrastructural details of the transfection process. Representative micrographs from three experiments and 100 observed sections. (
Footnotes
Acknowledgements
We are grateful to Peter Scherrer for reviewing the manuscript and to Marianne Vannauer for excellent technical assistance.