
Abstract
In plant systems, the green fluorescent protein (GFP) is increasingly used as a marker to study dynamics of the secretory apparatus using fluorescence microscopy. The purpose of this study was to immunogold localize the GFP, at the electron microscopic level, in a line of tobacco BY-2-cultured cells, expressing a GFP-tagged Golgi glycosyltransferase. To this end we have developed a simple, one-step chemical fixation method that allow good structural preservation and specific labeling with anti-GFP antibodies. Using this method, we have been able to show that an N-glycan GFP-tagged xylosyltransferase is specifically associated with Golgi stacks of BY-2 transformed cells and is preferentially located in medial cisternae. As an alternative to cryofixation methods, such as high-pressure freezing, which requires specialized and expensive equipment not available in most laboratories, this method offers researchers the opportunity to investigate GFP-tagged proteins of the endomembrane system in tobacco BY-2 cells.
I
A central issue for immunolocalization at the EM level is to preserve the epitope of interest while minimizing the loss of ultrastructural details. There are several strategies for the preparation of biological samples destined for immunogold labeling of proteins. These include (a) chemical fixation with aldehyde followed by progressive dehydration and embedding under low-temperature conditions (Carlemalm et al. 1982), (b) a brief chemical fixation with aldehyde followed by cryofixation and cryosectioning (Tokuyasu 1973), and (c) cryofixation techniques followed by freeze-substitution or freeze drying and embedding at low temperatures (Quintana 1994). The goals of these strategies are to stabilize the protein structure, retain protein antigenicity, and allow accessibility to antibodies. Compared to classical chemical fixation, the use of low temperature in cryomethods enhances the stability of biological material during solvent exchange and generates a minimal loss of macromolecular structures. Many of the cryotechniques are difficult to carry out in the majority of plant samples, which are more hydrated than animal specimens. There is, however, a general agreement that high-pressure freezing and freeze substitution (HPF/FS) provides the best preservation of cellular ultrastructures and antigenicity (Staehelin et al. 1990; Zhang and Staehelin 1992; Zhang et al. 1993; Driouich et al. 1993; Driouich and Staehelin 1997). Recently, the use of HPF/FS techniques has allowed the EM immunocytochemical localization of GFP in transgenic tobacco BY-2 cells (Nebenführ et al. 1999). However, this technique requires specialized and expensive equipment, which is not routinely available in most EM laboratories, whereas the need for localizing GFP-tagged proteins in transgenic plants is increasing. Therefore, it is important to develop alternative chemical fixation methods able to yield an optimal preservation of both the ultra-structures and antigenic sites. In this study we describe a one-step chemical fixation method for tobacco BY-2 cells, which consists of fixing cells in a mixture of glutaraldehyde and osmium at a low temperature. We show that this method allows an improved structural preservation of the endomembrane system and that the anti-GFP antibodies bind with high specificity, thereby allowing the localization of GFP-tagged glycosyltransferases within individual Golgi cisternae. The method can be easily applied in most laboratories without specialized equipment.
Materials and Methods
Growth Conditions
Cells were harvested by low-speed centrifugation (for 2 min) 3–5 days after subculturing and immediately used for experiments.
Construction of the XylT36::GFP and Transformation of Plant Cells
The fusion protein consisting of the first 36 amino acids of AtXylT (β1-2 xylosyltransferase from Arabidopsis thaliana) fused to the GFP (XylT36::GFP) was constructed as described in Pagny et al. (2003). The construct XylT36::GFP was used to transform Agrobacterium tumefaciens (LBA4404) (Höfgen and Willmitzer 1998). Transformation of tobacco BY-2 cells and initiation of suspension cultures of transgenic tobacco cells were performed as previously described (Gomord et al. 1998).
Production of Antibodies Directed Against GFP
The specific anti-GFP polyclonal antibodies were prepared in rabbits using commercial GFP (Q-biogen; Illkirch, France). The procedure of antibody preperation was as described in Faye et al. (1993).
Protein Extraction, SDS-PAGE, and Western blotting Analysis
Cell extracts were prepared as described in Gomord et al. (1997). Proteins were precipitated with TCA (12.5%) and solublized in denaturing buffer (SDS 3 × buffer; Pharmacia, Uppsala, Sweden). SDS-PAGE separation of proteins was performed according to Laemmli (1970). Electroblotting and immunodetection with the anti-GFP antibodies were carried out as previously described (Faye et al. 1993).
Fluorescence Microscopy
Five-day-old tobacco BY-2 cells expressing the fusion protein XylT36::GFP were observed using the standard fluorescein isothiocynate filter set. Image acquisitions were done with a Leica DMRB microscope coupled to an ORCA-ER CCD camera (C4742-95-12ER, Hamamatsu).
Sample Preparation for Electron Microscopy
Transgenic BY-2 cells expressing GFP-tagged Golgi glycosyltransferase were fixed in the dark for 30 min, 60 min, or 120 min at 4C in a solution consisting of 1% glutaraldehyde and 1% osmium tetroxide in 0.1 M Na cacodylate buffer, pH 7.2. This mixture was prepared and used immediately. After washing in distilled H2O, the samples were gradually dehydrated in 10%, 20%, and 40% aqueous ethanol (10 min in each bath), then in 60% and 80% (20 min in each bath), and finally in anhydrous ethanol for 30 min. After dehydration the samples were embedded in London Resin White (LRW) or Spurr resin. For the embedding in Spurr, cells were incubated twice in anhydrous acetone (5 min each), then infiltrated overnight at 4C in a mixture of Spurr and acetone (1/1) (v/v). Finally, the cells were bathed twice in pure and fresh resin (1 hr each). Polymerization was carried out at 60C for 24 hr. For the embedding in LRW, fixed and dehydrated cells were infiltrated at 4C in a mixture of LRW and ethanol, (1/2) (v/v) for 1.5 hr and (2/1) (v/v) for 1 hr. The cells were then incubated twice in pure resin overnight followed by 4 hr at 4C. Polymerization was carried out at 4C under UV light for 48 hr.
Electron Microscopy and Immunogold Labeling
Ultrastructural Observations. Ultrathin sections (80–90 nm) from Spurr and LRW-embedded samples were collected on formvar-coated copper grids and post-stained in 2% aqueous uranyl acetate and Reynold's lead citrate. Sections were examined at 80kV in a Philips electron microscope (Technai 12) at CCME (Rouen University; Rouen, France).
Detection of Complex N-glycans and Cell Wall Polysaccharides. The antibodies used were the rat monoclonal antibody JIM7 (Knox et al. 1990) specific for homogalacturonans, the rabbit polyclonal antibodies, anti-bupleuran, raised against a pectin polysaccharide of rhamnogalacturonan-I (RG-I) type (Sakurai et al. 1998), the rabbit polyclonal antibody anti-xyloglucan (Moore et al. 1986), and the purified polyclonal antibody specific for β1,2 xylose residues of plant N-glycans (Faye et al. 1993). The immunolabeling procedure of carbohydrate epitopes was as previously described (Zhang and Staehelin 1992; Vicré et al. 1998) with the following concentrations of antibodies: 1:20 for anti-xyloglucan, 1:10 for anti-β1,2 xylose, 1:100 for anti-bupleuran, and pure JIM7.
Immunolocalization of GFP. The anti-GFP antibody used in this study is a rabbit polyclonal serum produced in our laboratory. Formvar-coated 300-mesh nickel grids carrying ultrathin sections (90 nm) of LRW-embedded transgenic tobacco BY-2 were incubated in 0.5 M Nametaperiodate (NaIO4) for 30 min to remove excess fixatives. After washing in distilled H2O (twice for 10 min), grids were transferred to a 0.1 N HCl solution for 10 min to remove excess osmium tetroxide. Grids were then washed briefly and first blocked in 0.1 M glycine in PBS (0.01 M Na-phosphate, pH 7.2, and 0.15 M NaCl) containing 0.1% (v/v) Triton X-100 for 15 min to inactivate residual aldehydes. After washing in PBS containing 0.1% (v/v) Triton X-100 for 5 min, sections were blocked with the second blocking buffer solution consisting of normal goat serum (British Biocell; Cardiff, UK) 1:30 in PBS containing 0.1% (w/v) BSA and 0.2% (v/v) Tween-20 for 45 min. Grids were then incubated in the primary anti-GFP antibodies diluted 1:100 in the second blocking buffer at 4C overnight. A control was performed under the same conditions with the rabbit preimmune serum diluted 1:100 in the first blocking buffer solution. The grids were then washed once in PBS containing 0.1% (v/v) Triton X-100 for 5 min, then twice in TBS (0.05 M Tris-HCl, pH 8.4, and 0.15 M NaCl) containing 0.1% (v/v) Triton X-100 for 15 min each, and blocked for 45 min with the third blocking buffer solution consisting of normal goat serum (British Biocell) 1:30 in TBS containing 1% (w/v) BSA, 0.1% (v/v) Triton X-100, and 0.2% (v/v) Tween-20. The sections were then transferred to a droplet of goat anti-rabbit IgG conjugated to 10-nm gold particles (British Biocell) diluted 1:25 in the third blocking buffer solution for 2 hr at room temperature. After three washes in PBS containing 0.1% (v/v) Triton X-100 for 20 min and two washes in distilled H2O for 20 min, grids were finally post-stained and examined as described above.
Results
Ultrastructural Features of Tobacco BY-2 Cells
We first examined the ultrastructural features of tobacco BY-2 cells co-fixed with a glutaraldehyde/osmium mixture and embedded in Spurr resin (Figure 1). After 30 min of fixation, most if not all cell compartments were well preserved. The endomembranes, including nuclear envelope, the plasma membrane, tonoplast, Golgi stacks, and the endoplasmic reticulum (ER) were consistently discernable (Figures 1A and 1B). A slight swelling of the ER, which is a common phenomenon of fixation, was observed at different times of fixation (Figures 1A 1C, and 1E). In the context of this article, it is worth noting that the organization of Golgi stacks was typical of plant suspension-cultured cells (Zhang and Staehelin 1992; Driouich et al. 1993). As seen in Figure 1D, different subtypes of Golgi cisternae, cis, medial, and trans, can be identified on the basis of their staining patterns as characterized earlier (Staehelin et al. 1990; Zhang and Staehelin 1992). It is also worth noting that cortical microtubules were well preserved and were frequently more visible at fixation times of 60 min or more (Figures 1F and 1G).
We also examined the ultrastructure of the same cells embedded in LRW resin, which is more amenable to immunogold labeling. In LRW-embedded cells, although some cytoplasmic extraction was observed, Golgi stacks and ER were quite well preserved and clearly distinguishable. As illustrated in Figure 2, even though the quality of structural preservation did not match that of the Spurr-embedded cells (Figure 1 and 2B), the cis-to-trans polarity of Golgi stacks as well as subtypes of Golgi cisternae were identifiable. In most cases, the best preservation of these Golgi structures in LRW-embedded samples was obtained at a fixation time of 120 min (Figure 2A) vs 60 or 30 min of fixation.
Immunolocalization of Complex N-glycans and Cell Wall Polysaccharides
Before the immunolocalization of GFP, we checked whether the co-fixation protocol allowed classical immunogold labeling of plant cell wall polysaccharides and N-glycans both in the cell wall and Golgi stacks of tobacco BY-2 cells. As shown in Figures 3A and 3B, specific and heavy immunogold labeling was found over Golgi cisternae and the cell wall using antibodies directed either against xyloglucan, the major hemicellulose polysaccharide in primary walls of dicots (Moore et al. 1986), or against β1-2 xylose residues of plant N-glycans (Faye et al. 1993). Similarly, anti-bupleuran antibodies raised against a pectin polysaccharide of RG-I type labeled Golgi cisternae as well as the entire cell wall (Figure 3C). The MAb JIM7, which also recognizes pectins, showed sparse labeling of the cell wall (Figure 3D) and Golgi stacks (not shown). These results demonstrate that the co-fixation method is suitable for immunogold localization of sugar epitopes associated with either N-glycans or cell wall matrix polysaccharides.
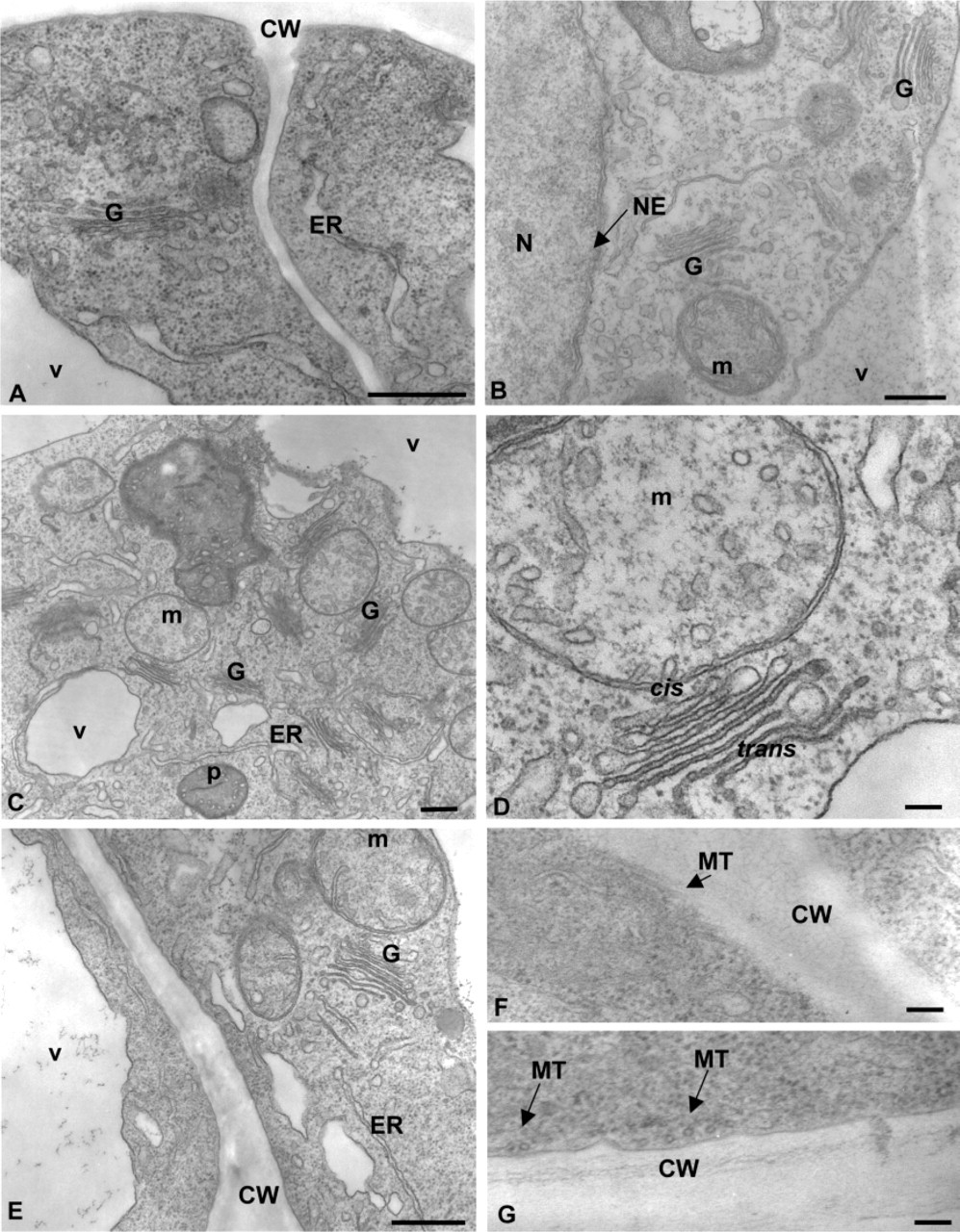
Electron micrographs of transformed suspension-cultured tobacco BY-2 cells preserved by the co-fixation technique. Cells were fixed for 30 (
Immunolocalization of Golgi-targeted GFP
Specificity of the Antibodies. Before investigating the immunogold localization of GFP on transformed tobacco BY-2 cells, we checked for the specificity of anti-GFP antibodies that we had produced in our laboratory, using immunoblotting technique. A total extract of intracellular proteins from non-transformed and transformed suspension-cultured BY-2 tobacco cells and a purified GFP, which was used for immunization, were probed on a Western blot with anti-GFP antibodies. As shown in Figure 4, the purified GFP (∼29 kD), was also detected with the anti-GFP antibodies (Figure 4, Lane 1). Although no signal was obtained with the protein extract of wild-type tobacco cells (Figure 4, Lane 2), anti-GFP antibodies detected a major polypeptide in the intracellular fraction of transformed cells (Figure 4, Lane 3). This polypeptide, of ∼32 kD, corresponds to the fusion protein AtXylT36::GFP. No immunostaining was observed with the preimmune control serum (not shown). These data demonstrate the specificity of anti-GFP antibodies.
Immunogold Localization of GFP. To investigate the immunogold localization of GFP, sections of transformed LRW-embedded BY-2 cells were immunolabeled with antibodies directed against GFP. We first performed and quantified the immunogold labeling over Golgi cisternae before and after a treatment with NaIO4 and HCl. The data are presented as a histogram in Figure 5. In untreated samples, binding of anti-GFP antibodies to the Golgi cisternae was weak, with most of the stacks carrying about three gold particles. In contrast, after NaIO4/HCl treatment, a much heavier labeling, four- to sevenfold higher than in untreated sections, was seen over the Golgi stacks. This demonstrates that the treatment was necessary to achieve significant and specific labeling of GFP within Golgi stacks in LRW-embedded cells. In addition, the density of GFP labeling seemed to be affected by the duration of fixation (Figure 5), with the highest density obtained with 60 min-fixed samples. When Spurr-embedded samples were used, GFP immunogold labeling was difficult to achieve and no significant labeling was obtained even after treatment with NaIO4 and HCl (data not shown).
Figure 6B illustrates the typical immunogold labeling pattern obtained with anti-GFP antibodies on LRW-embedded transformed BY-2 cells. Labeling was mostly found over the medial cisternae, with less over the cis and trans cisternae. The ER, tonoplast, and the vacuole were never labeled (Figure 6B). Scattered and very weak labeling was found over the plasma membrane and the cell wall with both anti-GFP and preimmune sera. Finally, no significant labeling was found in control experiments performed either using preimmune serum (Figure 5) or with the omission of the primary antibodies (not shown).
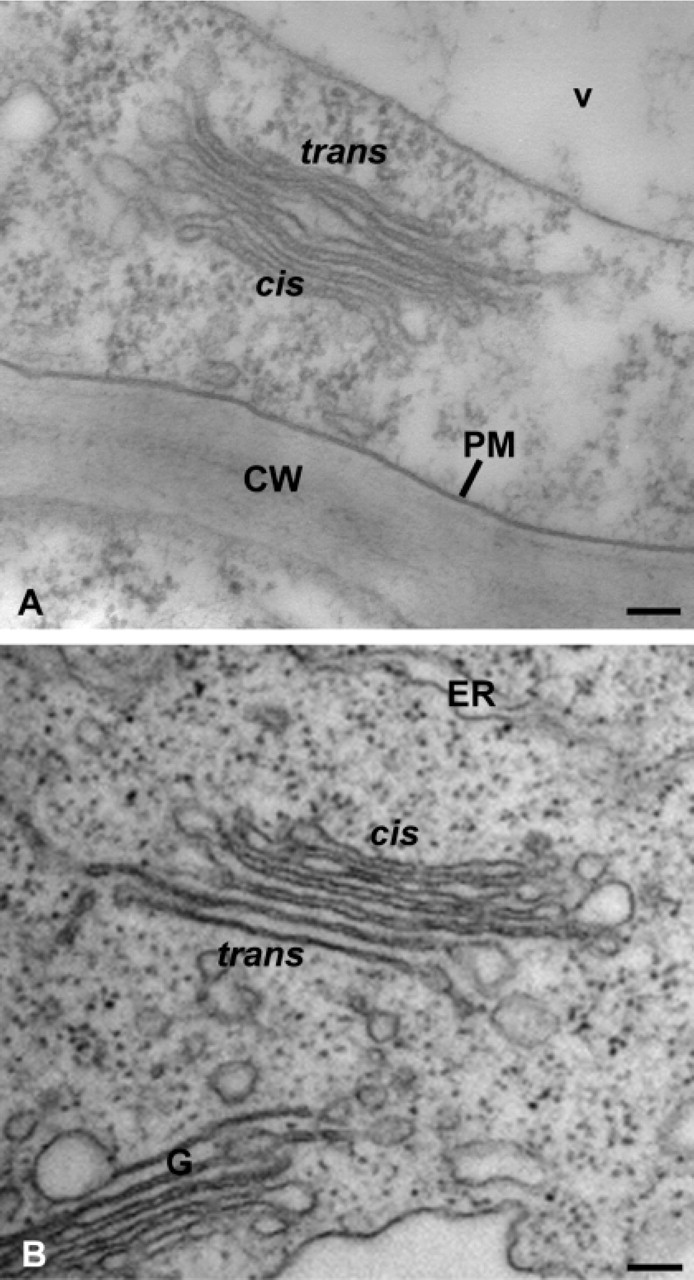
Electron micrographs of Golgi stacks in transformed suspension-cultured tobacco BY-2 cells preserved by the co-fixation technique. Cells were embedded either in LRW (
Together, the immunogold labeling results confirm that the punctate pattern of GFP fluorescence in BY-2 cells shown in Figure 6A corresponds indeed to dispersed individual Golgi stacks and that the fusion protein was targeted to the Golgi apparatus. In addition, quantitative analysis of the binding pattern of GFP demonstrates that the β1,2 xylosyltranferase involved in N-glycan processing is mostly located in medial cisternae of Golgi stacks in tobacco BY-2 cells (see also Pagny et al. 2003).
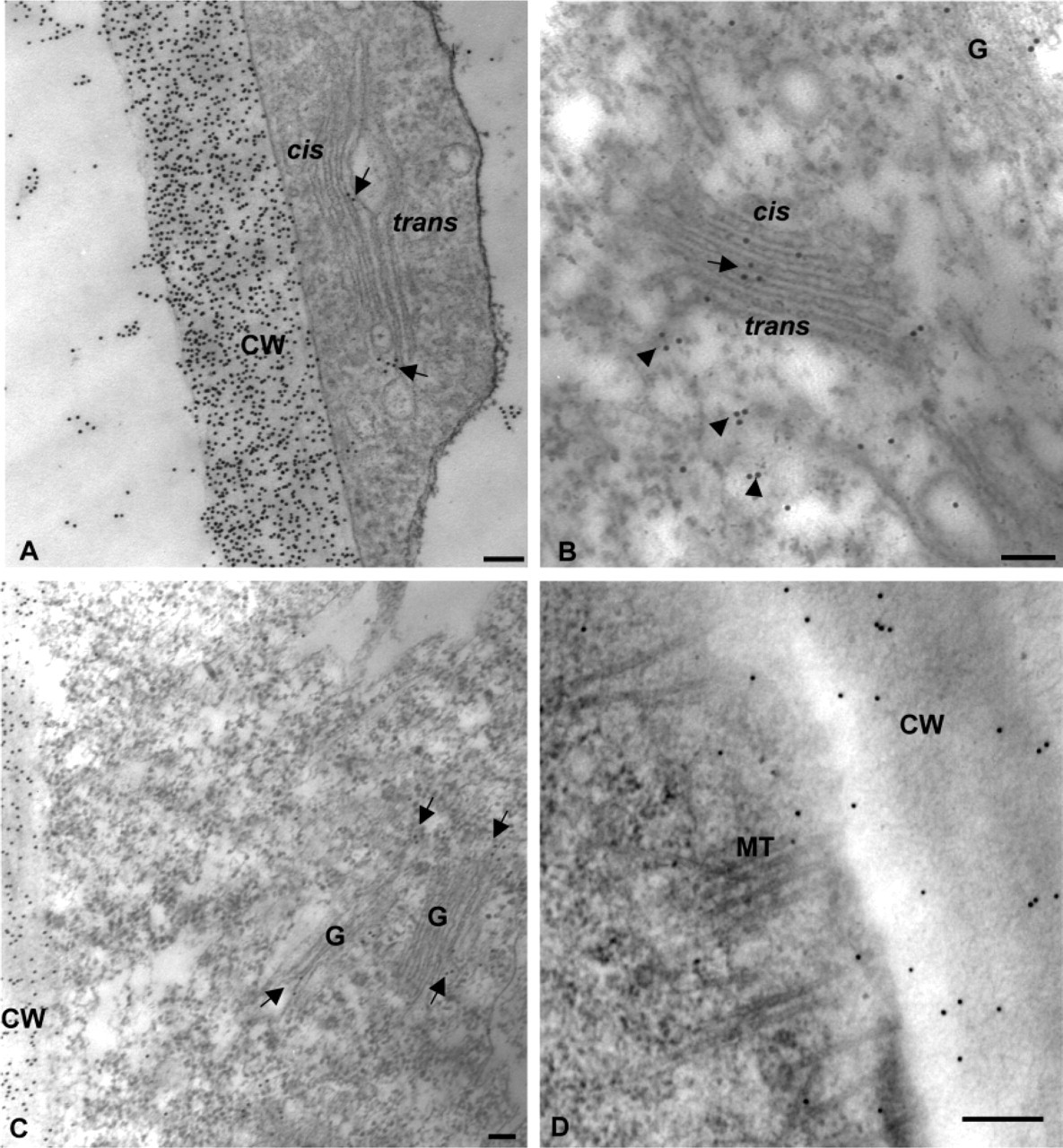
Immunogold labeling of Golgi stacks and cell walls with anti-polysaccharide and anti-N-glycan antibodies. (
Discussion
Fixation Protocols and Ultrasructural Preservation
Many chemical fixation methods are currently used to prepare specimens for immunogold labeling of proteins at the EM level (Robinson et al. 1994; Frigerio et al. 2000; Nikus et al. 2001). They usually consist of fixing samples with 4% (w/v) paraformaldehyde and 0.25–2% (w/v) glutaraldehyde eventually followed by a postfixation with 1% (w/v) osmium, before dehydration and embedding in LRW resin. When applied to tobacco BY-2 suspension-cultured cells, these protocols yield cells that are not structurally well preserved. In particular, important extractions of the ground cytoplasm and very poor preserved endomembranes, including Golgi stacks, were consistently observed. Therefore, the samples were not used further for immunogold labeling experiments with GFP. We therefore performed an uncommon process consisting of fixing tobacco cells in only a one-step using a mixture of glutaraldehyde and osmium (i.e., a co-fixation) at 4C. A similar method, but including an additional post-osmication step, has been used to explore the ultrastructures of algae, fungi, stems and leaves of higher plants, and isolated subcellular fractions (Hayat 2000). Our modified method provided a good structural preservation of suspension-cultured tobacco BY-2 cells at all times of fixation (see Figure 1). In particular, Golgi stacks were well preserved and easily discernable. The quality of preservation of Golgi morphology did not match that obtained in high pressure-frozen tobacco BY-2 cells (Winicur et al. 1998; Nebenführ et al. 1999), but was in most cases better than obtained in tobacco BY-2 cells (Wee et al. 1998; Fitchette et al. 1999) or other plant cells, including carrot suspension cultures, clover, and flax roots (Moore et al. 1991; Lynch and Staehelin 1992; Vicré et al. 1998) prepared by classical chemical methods. Golgi stacks were, however, less distinct in LRW-embedded cells than in Spurr-embedded samples, which is commonly observed for all plant and animal cells when LRW is used. The fixation time of 120 min was necessary in our conditions to obtain a sufficient preservation of LRW-embedded cells and discernable Golgi cisternae (Figure 2). It is worth noting that cortical microtubules, which are reputed to be difficult to preserve even with some cryomethods, were distinguishable in most cells. We discuss below that the method can be well exploited for immunogold localization of GFP within Golgi stacks.

Specificity of anti-GFP antibodies. Commercial GFP (Lane 1) or cell proteins from wild-type (Lane 2) or transformed suspension-cultured tobacco BY-2 cells expressing the recombinant XylT36::GFP (Lane 3) were separated by SDS-PAGE, transferred onto nitrocellulose, and probed with the anti-GFP antibodies. Arrowheads at left indicate the position of the molecular mass markers (14, 18, 29, 43, 68, and 94 kD from bottom to top).
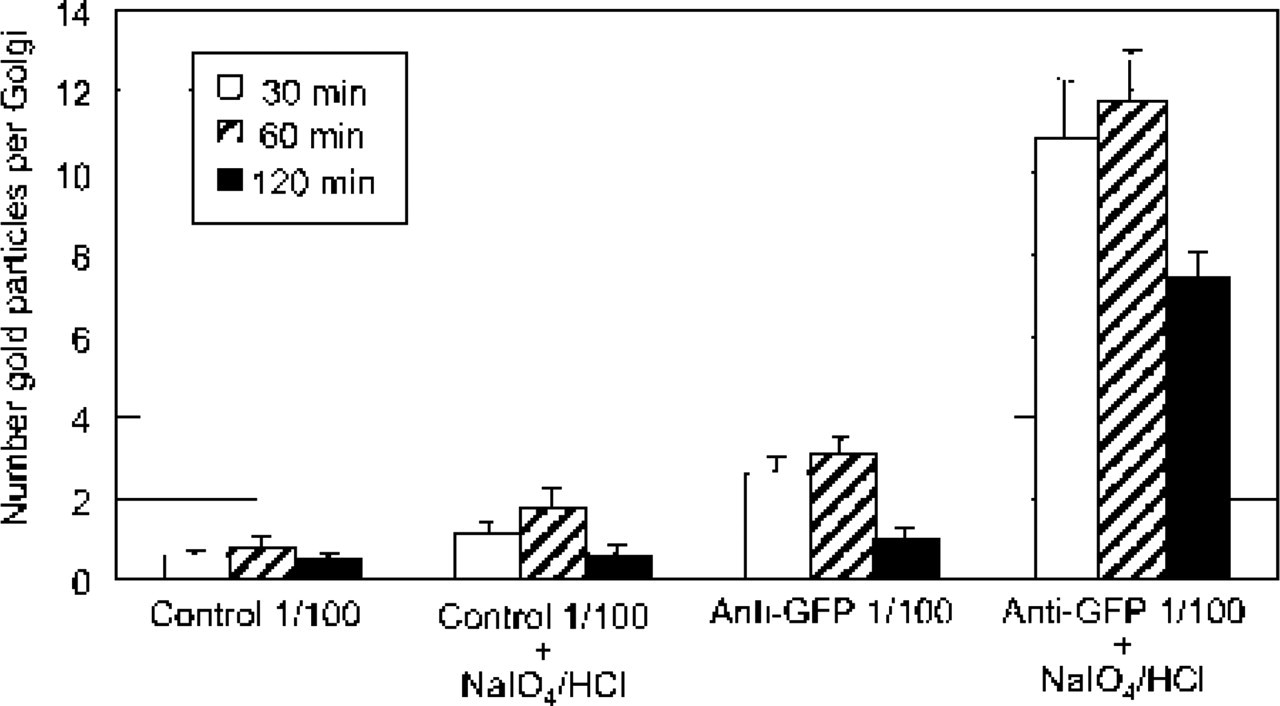
Histogram illustrating the labeling pattern of Golgi stacks with the anti-GFP antibodies in transformed suspension-cultured tobacco BY-2 cells at different times of fixation and with or without an etching treatment (NaIO4/HCl). Anti-GFP labeling was carried out on LRW-embedded cells. Highly specific labeling of Golgi stacks is obtained after etching. Each histogram represents the mean of 30 values ± SEM. Control 1/100, rabbit preimmune serum used at a dilution of 1:100. Anti-GFP 1/100, polyclonal anti-GFP antibodies used at a dilution of 1:100.
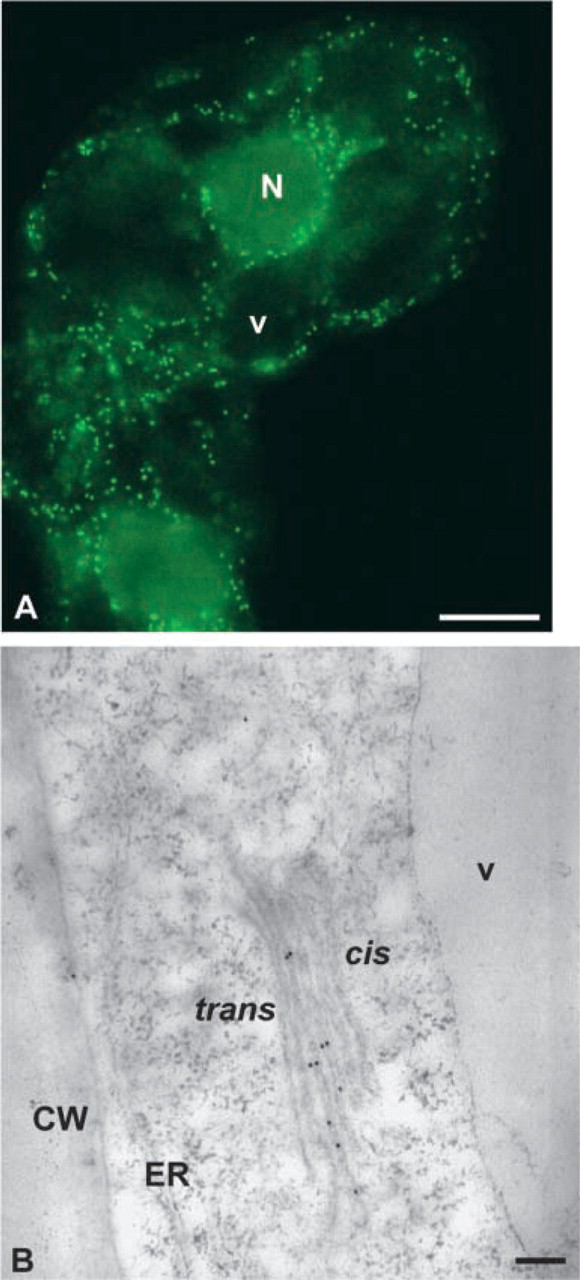
Distribution of XylT36::GFP in transgenic tobacco BY-2 suspension-cultured cells. (
Immunogold Labeling of Golgi-targeted GFP
We demonstrate that antibodies recognizing both carbohydrate epitopes and GFP bind to Golgi stacks of tobacco BY-2 cells prepared by the co-fixation method. First, immunogold labeling of cell matrix polysaccharides and complex N-glycans was possible and the quality of Golgi labeling with anti-β1-2 xylose, anti-xyloglucan, and anti-pectin antibodies, did not differ from those previously observed in many plant cells either chemically or cryofixed (Moore et al. 1991; Lynch and Staehelin 1992; Zhang and Staehelin 1992; Driouich et al. 1993; Vicré et al. 1998). However, carbohydrate antigens are known to be less sensitive to fixative crosslinking than proteins, which probably facilitated polysaccharide antibody binding. Second, a specific GFP localization was also achieved in well-distinguishable Golgi stacks. As to immunocytochemical detection of proteins, it is known that glutaraldehyde crosslinks the amino groups of proteins and osmium causes conformational changes, thus reducing protein antigenicity. However, it has been reported that glutaraldehyde used alone is more effective in crosslinking proteins than when used in combination with osmium and that osmium induces deformation of protein structures when the specimen is prefixed with glutaraldehyde (Hayat 2000). Therefore, the use of the co-fixation could be more favorable to immunogold labeling of proteins than double classical fixation techniques. However, we did not check this statement in our case because no GFP labeling was done on the BY-2 cells fixed with the conventional procedure because of the very poor ultrastructural preservation. The binding of GFP antibodies to Golgi cisternae in co-fixed BY-2 cells was specific whatever the duration of fixation (Figure 5). However, this necessitates a treatement with NaIO4 and HCl before immunolabeling, known to eliminate bound osmium (Craig and Goodchild 1984; Brorson 1998) and to reduce imine bonds between glutaraldehyde and proteins (Hayat 1970). The drawback of this treatment is the potential loss of structural details of the membranes. In our conditions, this always resulted in a higher specific labeling, probably due to the unmasking of antigenic determinants and enhanced accessibility of the antibodies to GFP. It had, however, little influence on the ultrastructure and contrast of Golgi stacks, still allowing subtypes of Golgi cisternae and intra-Golgi distribution of gold particles to be discernable (see Figure 6). Although variability occurred from one experiment to another, the increase in GFP binding upon treatment was four- to sevenfold greater depending on the fixation time, and was even much higher than the non-specific binding of preimmune serum (see controls in Figure 5). The most significant binding with the least background (preimmune labeling) was obtained at a fixation time of 120 min, probably due to increased stabilization of GFP-tagged glycosyltransferase within Golgi structures.
The glycosyltransferase fused to GFP is a β1,2 xylosyltransferase from Arabidopsis thaliana, which is a type II membrane protein located, along with all other glycosyltransferases involved in N-glycan processing and complex polysaccharide synthesis, in Golgi membranes (Keegstra and Raikhel 2001). This enzyme is involved in the addition of β1,2 xylose to N-linked glycoproteins within Golgi stacks of plants cells, an event shown to occur predominantly in medial cisternae (Lainé et al. 1991; Zhang and Staehelin 1992). In this study, we found that the β1,2 xylosyltransferase was mainly confined to medial cisternae (see also Pagny et al. 2003), which is the same Golgi location of its products (i.e., β1,2 xylose containing N-linked glycoproteins). This demonstrates that the enzyme acts preferentially in medial cisternae to add xylose residues onto N-glycoproteins and that glycosyltransferases involved in N-glycan processing are at least partially compartmentalized within Golgi stacks in plant cells.
In summary, we developed a simple co-fixation method of tobacco BY-2 suspension-cultured cells that provides a good preservation of cell ultrastructure while maintaining antigenicity of complex polysaccharides, N-glycans, and proteins of the Golgi membranes. We also show that the co-fixation method allows a specific localization of Golgi-targeted GFP using immunogold EM and NaIO4/HCl etching treatment. Although the HPF/FS remains the best technique for such purposes, the chemical co-fixation and immunocytochemical methods described here can be easily used to study GFP-tagged plant proteins of the endomembrane system in laboratories for which specialized HPF equipment is not available.
Footnotes
Acknowledgements
Supported by the CNRS and the University of Rouen. We are grateful to Dr Anja Geitman (University of Laval), to Dr Thomas Giddings (University of Colorado), and to John Moore (University of Cape Town) for helpful comments and critical reading of the manuscript. Thanks are also due to Laurence Chevalier and Jean Herbet for technical assistance and help at the Centre Commun de Microscopie Electronique (Rouen University).