
Abstract
Macrophage migration inhibitory factor (MIF) is a key mediator in inflammatory or immune-mediated diseases, although its role in heart diseases is unknown. This study investigated the expression of MIF in the myocardium in the development of acute myocardial infarction (AMI). By use of immunohistochemistry, Western blotting, RT-PCR, and in situ hybridization, the gene and protein expression of MIF in the heart at 6 hr, 1 day, 3 days, 1 week, and 2 weeks after AMI was studied. In both normal and sham-operated rats, MIF mRNA and protein were expressed constitutively at low levels by the myocytes. By contrast, MIF mRNA was rapidly upregulated by the surviving myocytes in the infarcted region and, to a lesser extent, the non-infarcted region, accounting for a sevenfold increase at 6 hr after AMI (p<0.001). This was followed by a fourfold increase in MIF protein expression at day 1 after AMI (p<0.05). Macrophages were found accumulated in the infarcted region, being significant at day 1 (p<0.01) and progressive increased over the 2-week time course (p<0.01) in which MIF was found expressed in these cells. The results indicated that the infiltrating macrophages and myocytes were sources of MIF in the infarcted region. The latter cells became activated and involved in the amplification of inflammatory response in AMI. Therefore, upregulation of myocardial MIF may contribute to macrophage accumulation in the infarcted region and their pro-inflammatory role may participate in the myocyte damage seen in AMI.
Keywords
M
Recently, we have demonstrated that, in patients with acute myocardial infarction (AMI), MIF levels were significantly elevated and were higher than those in patients with reversible myocardial ischemia (Yu et al. 2001a). Because the process of myocyte necrosis in AMI involves the induction of an inflammatory response and subsequent infiltration of inflammatory cells (Yu et al. 2001b), we postulate that MIF, as a key proinflammatory cytokine, may participate in the development of AMI, which was examined in the present study.
Materials and Methods
Induction of AMI in Rats
Left coronary artery occlusion was performed in 12-week male Sprague-Dawley rats as previously described (Fishbein et al. 1978; Yu et al. 2001b). Anesthetized rats were intubated and ventilated, and left thoracotomy was performed at the fourth and fifth ribs. The left anterior descending artery was ligated 2 mm from the origin of the aorta with 6.0 silk. Sham-operated rats underwent the same operation but without ligating the coronary artery. Rats were sacrificed at 6 hr, 1 day, 3 days, 1 week, or 2 weeks after the surgical procedure. The investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996).
In Situ Hybridization
A 420-bp Xba I/BamH I fragment of mouse MIF cDNA in the Bluescript SK+ vector (Stratagene; La Jolla, CA), which is highly homologous to the rat sequence (Sakai et al. 1994; Lin et al. 2000), was used to prepare sense and antisense digoxigenin-11-UTP MIF cRNA probes according to the manufacturer's protocol (Roche; Mannheim, Germany). In situ hybridization was performed on 4-μm formalin-fixed, paraffin-embedded sections by a microwave heating technique (Lan et al. 1995,1996). Sections of the heart for in situ hybridization underwent morphometric measurement by systematically scanning the regions using a Zeiss Axiophot microscope at a magnification of X200 (Yu et al. 2001b). Each image was analyzed using hue, saturation, and intensity detection mode of Leica Qwin Image Analyzer (Leica; Wetzlar, Germany) under a standardized brightness and contrast of light source according to the user manual. Mean values of at least 15 fields were taken in each region of the heart.
Immunohistochemistry
Double immunostaining of MIF expression by infiltrating macrophages in infarcted or non-infarcted heart tissues was performed on 4-μm paraffin sections of formalin-fixed heart with a microwave-based, multiple immunoenzymatic staining (Lan et al. 1995) using monoclonal anti-MIF (III.D.9 IgG) and mouse monoclonal anti-ED-1 antibodies as previously described (Serotec; Oxford, England) (Lin et al. 2000; Yu et al. 2001b).
Western Blotting Analysis and EIA Detection of Plasma MIF Level
Total cellular protein was extracted by 50 mM Tris-HCl, pH 8, 100 mM NaCl, 1% Triton X-100, 5 mM EDTA, 1 mM PMSF and a mini-complete proteinase inhibitor cocktail (Roche). Western blotting of MIF has been previously described (Lan et al. 1997; Lin et al. 2000). Positive signals were then visualized by an enhanced chemiluminescence (ECL) detection system (Amersham-Pharmacia; Arlington Heights, IL). Plasma levels of MIF were measured with a ChemiKine Rat/Mouse MIF EIA kit (Chemicon; Temecula, CA).
Reverse Transcription Polymerase Chain Reaction (RT-PCR)
Frozen heart tissue samples were homogenized and RNA was extracted by the acid-phenol-guanidinium-thiocyanate method (Chomczynski and Cantin 1987). The expression of MIF, IL-1β, and TNF-α mRNA was detected by RT-PCR using TATA-box binding protein (TBP) as an internal control. The primers used were MIF, 5’ CCA GGA CCG CAA CTA CAG CAA 3’ (forward) and 5’ GGG CTC AAG CGA AGG TGG AAC 3’ (reverse); IL-1β, 5’ CCT TCT TTT CCT TCA TCT TTG 3’ (forward) and 5’ ACC GCT TTT CCA TCT TCT TCT 3’ (reverse); TNF-α, 5’ CGT CGT AGC AAA CCA CCA AGC 3'; and TBP, 5’ ACC CTT CAC CAA TGA CTC CTA TG 3’ (forward) and 5’ ATG ATG ACT GCA GCA AAT CGC 3’ (reverse). PCR was performed at 94C for 45 sec, 58C for 40 sec, and 72C for 45 sec for 29 cycles. Validations of the semiquantitative RT-PCR were performed using normal rat left ventricular cDNA to ensure that there was no interaction between two pairs of primers, and the reaction condition was standardized for different amounts of cDNA (Jonas et al. 1999).
Statistical Analysis
The differences in mean value among the various rat groups were analyzed by one-way analysis of variance (ANOVA) with Scheffe's correction or paired sample t-test where appropriate, using a statistical software program (SPSS for Windows, ver. 10.0; SPSS, Chicago, IL). All data were expressed as mean ± SD. A p value <0.05 was considered statistically significant.
Results
The systolic and diastolic blood pressure remained normal and did not differ between sham and AMI animals throughout the experiments (systolic: sham, 113 ± 12 vs 107 ± 13 mmHg, p = NS; diastolic, sham 93 ± 14 vs 88 ± 13 mmHg, p = NS). There was no difference in the body weight between the groups over the 2-week time course (sham 395 ± 48 vs 411 ± 50 g, p = NS).
Myocardial MIF mRNA and Protein Expression in Normal Hearts and Animals with AMI
Weak MIF mRNA and protein were constitutively expressed by myocardium in both normal and sham-operated rats (Figures 1a, 1b, 2a, 2b, 3, and 4). In contrast, by in situ hybridization and RT-PCR, MIF mRNA was markedly expressed by myocytes in the infarcted region, peaking at 6 hr after AMI (p < 0.001) (Figures 1c and 3; Table 1). Upregulation of MIF mRNA by myocytes remained high throughout the study period (Figures 1c-1g). In the noninfarcted area, myocardial MIF mRNA was also upregulated (p<0.01), although to a lesser extent compared to the infarcted region (Figures 1h and 3; Table 1). The MIF protein expression in the infarcted region did not show an early peak but increased gradually over the 2-week study period (Figures 2 and 4; Table 1). Meanwhile, ELISA showed that the MIF level in serum was significantly increased at 6 hr (p<0.001) (Table 1). This may reflect a rapid secretion of the stored cytoplasmic MIF from the damaged myocytes after AMI.
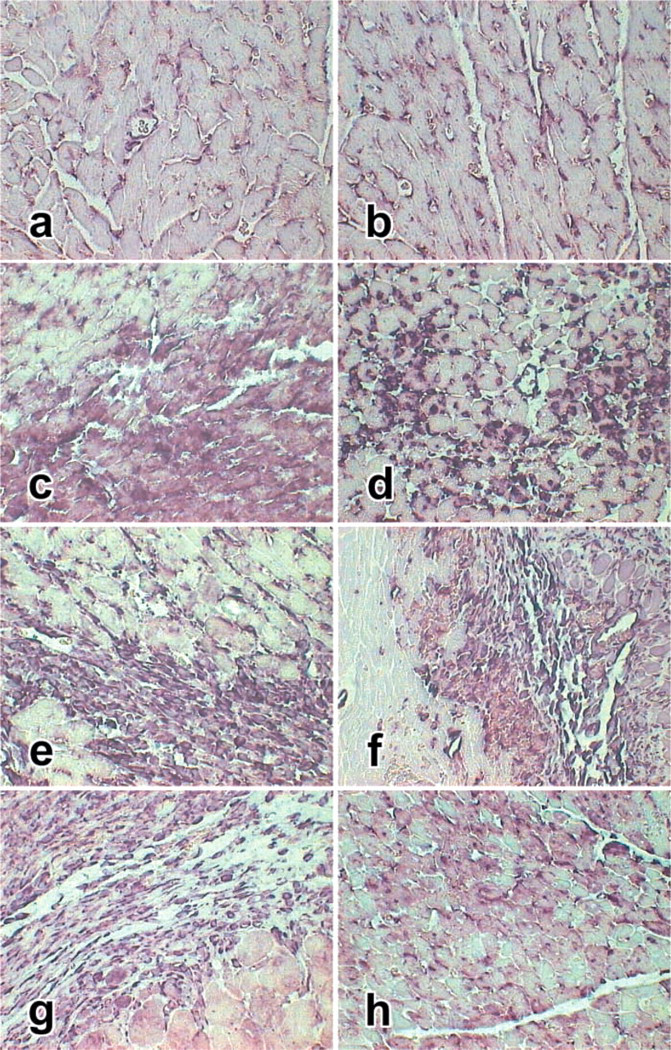
MIF mRNA expression demonstrated by in situ hybridization. The MIF mRNA was stained purple. (
Myocardial MIF Expression and Macrophage Accumulation in the Infarcted Region
As shown in Figure 2, there was no macrophage infiltration in the normal, sham-operated, and noninfarcted heart tissues. In contrast, a significant increase in the number of macrophages was seen at day 1 (p<0.05) and continued to increase throughout the experimental time course (p<0.001) (Figures 2d-2g, Table 1). Infiltrating macrophages were localized exclusively within the infarcted region, where strong MIF expression was found.
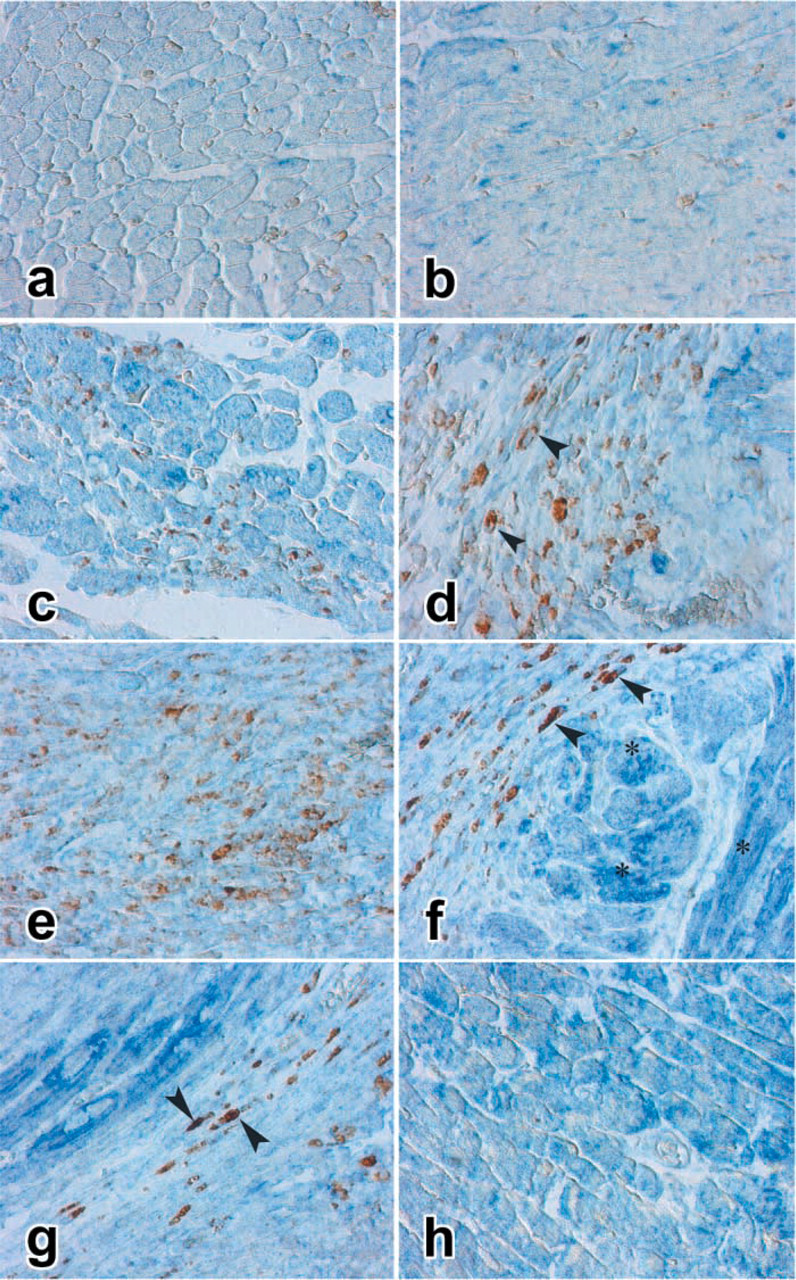
Double immunostaining of MIF (blue) and macrophages (brown) in hearts with AMI. (
MIF Expression by Infiltrating Macrophages
Marked upregulation of MIF mRNA and protein was also found in infiltrating macrophages in the infarcted region. As shown in Figure 1, strong MIF mRNA was found in the macrophages in the infarcted region compared to myocytes in the noninfarcted area (Figures 1d-1h). This was further confirmed by double immunostaining, showing that the ED1+ macrophages were co-expressing MIF within the infarct region (Figures 2d-2g, Table 1), indicating that the activated macrophages are also the major source of MIF during AMI. Correlation analysis showed that upregulation of myocardial MIF correlated closely with the number of infiltrating macrophages in the infarcted lesions (r = 0.61; p<0.01).
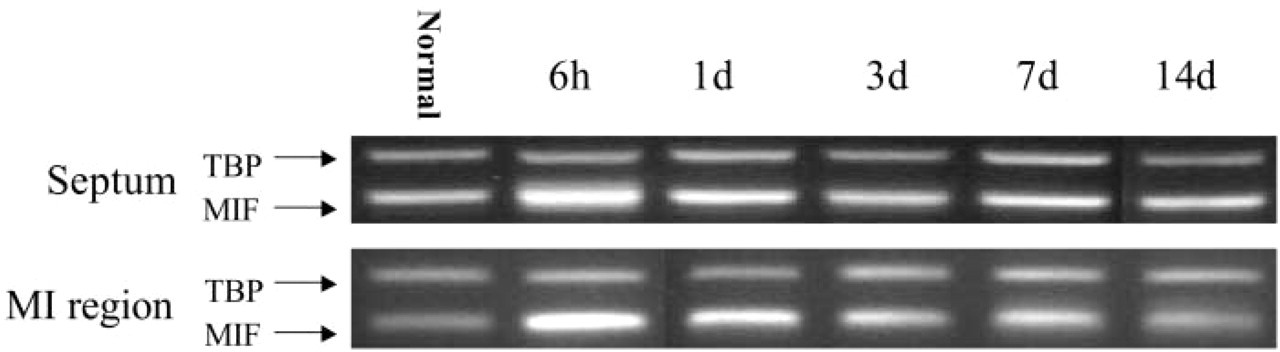
Multiplex RT-PCR demonstrated the MIF mRNA expression in hearts with AMI. TBP was used as an internal control. The relative abundance of MIF mRNA is expressed as the fraction of internal control and is summarized in Table 1. The MIF mRNA levels peaked at 6 hr and then decreased gradually over 2 weeks. The infarcted region in the normal represents sham-operated rats. n = 5 per group.
Expression of Interleukin-1β and Tumor Necrosis Factor-α in Normal Hearts and Animals with AMI
In the infarcted region, IL-1β level was significantly elevated by more than ninefold in the first day of AMI (p<0.01) (Table 1). The level decreased very rapidly afterwards and was no different from the control level from 3 days to 2 weeks. A similar, although smaller surge of IL-1β level was observed in the non-infarcted area on day 1 (p<0.05), which decreased afterwards. For TNF-α level, it followed a very similar time course of change to that of IL-1β, becoming elevated by >20-fold on day 1 in the infarcted region (p<0.001) and decreasing rapidly thereafter (Table 1). In the noninfarcted region, similar changes were observed, although to a much lesser extent than that of the infarcted region.
Discussion
This study demonstrated, for the first time, that upregulation of MIF is involved in AMI. In normal heart, the pre-formed MIF was constitutively and weakly expressed by myocytes. After AMI, MIF mRNA was rapidly upregulated in myocytes within hours, preceding the macrophage infiltration in the infarcted region on day 1. The increase in serum MIF level after AMI and loss of cytoplasmic MIF protein in myocytes suggests that MIF may be rapidly secreted from damaged myocytes. Alternatively, ischemic myocytes may not be able to synthesize MIF protein which, in general, is a later process than mRNA synthesis. These may explain a late and smaller peak observed for protein expression in the infarcted region. On the other hand, the elevated serum MIF level may also be a result of systemic response, although it was not observed in patients with unstable angina in a previous study (Yu et al. 2001a). The early and rapid upregulation and secretion of MIF by myocytes in the infarcted region, followed by an active inflammatory response including abundant macrophage accumulation and activation, indicated that MIF may play a role in augmenting inflammation-mediated myocardial injury in AMI. On the other hand, cytoplasmic MIF protein and mRNA in the noninfarcted region of the myocardium remained high in the early course of AMI. When located in the peri-infarct zone, it may further amplify the inflammatory response in the infarct zone.
Macrophages, which had originally been considered to be the target of MIF action, were identified as one of the major sources of MIF production during AMI, being evident from day 1 onwards. Indeed, almost all infiltrating macrophages expressed MIF in the infarcted region. Macrophage-derived MIF may both initiate and amplify the inflammatory process during AMI by recruiting more macrophages, neutrophils, and T-cells to the site of inflammation by upregulating the adhesion molecules such as ICAM-1 (Huang et al. 2001), by mediating the removal of damaged or necrotic myocytes, by promoting the expression of additional proinflammatory mediators, such as inducible nitric oxide synthase (Calandra et al. 1994,1995), and consequently contributing to the remodeling process of the infarcted heart. In the current study, increase in gene expression of other inflammatory cytokines was also observed within the first day of AMI, including IL-1β and TNF-α. Such changes were more dramatic in the infarcted region, where myocyte damage was obvious after cellular hypoxia.
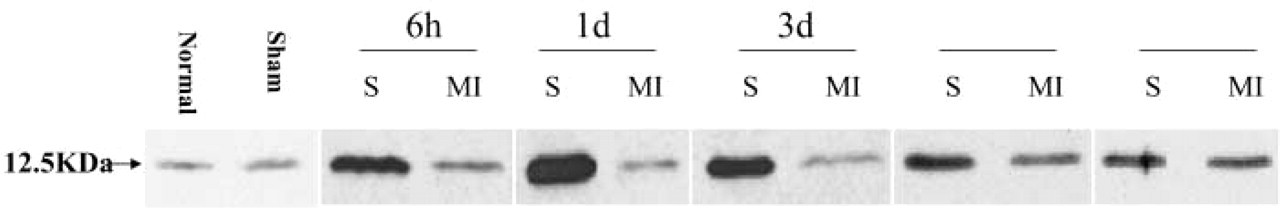
Western blotting analysis of MIF at different time points and regions of hearts with AMI. The MIF protein was detected in normal rat heart at a low level. After AMI, MIF was markedly increased in the noninfarcted septum at 1 day and continues to be expressed over 2 weeks. However, in the infarcted region the MIF level was lower than in the noninfarcted region throughout the study. The densitometric values of MIF protein are expressed as the fraction of recombinant MIF control. Sep, noninfarcted septum; MI, infarcted region. n = 5 per group.
Expression of macrophage migration inhibitory factor (MIF) protein and mRNA as well as interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) gene expression in the infarcted zone (MI) and the noninfarcted septum in rats (n = 5–6 in each group) a
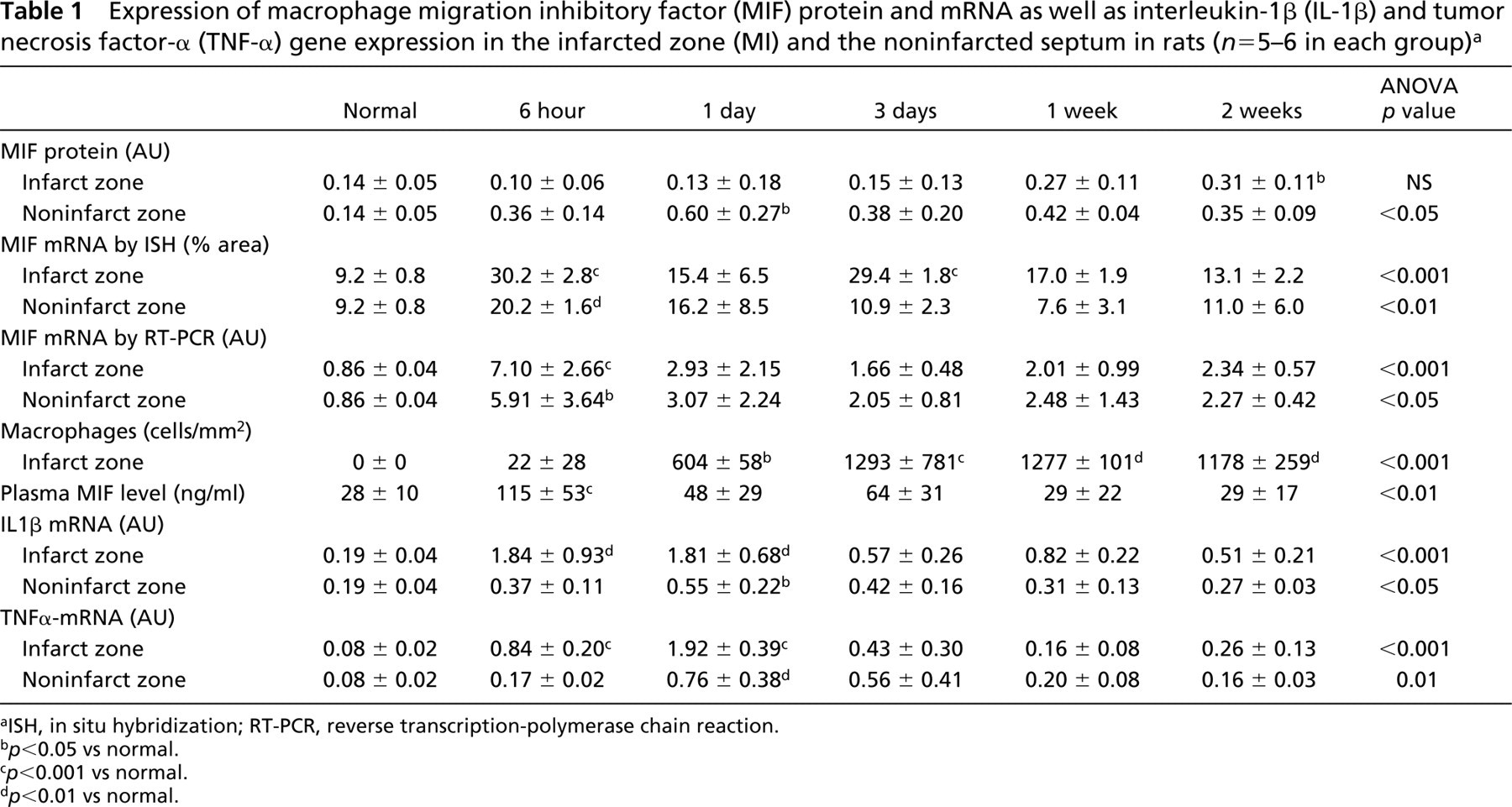
aISH, in situ hybridization; RT-PCR, reverse transcription-polymerase chain reaction.
b p<0.05 vs normal.
c p<0.001 vs normal.
d p<0.01 vs normal.
It has been shown that myocyte damage after AMI is mediated by an inflammation-like response. This was supported by the observation in animal studies that infiltration of inflammatory cells, such as macrophages and neutrophils, in the infarct zone was evident early after AMI (Sun et al. 1994; Yu et al. 2001b). The present study further suggests the role of an inflammatory response in AMI, which is mediated through MIF in the event of irreversible myocardial ischemia. MIF has been shown to be a key cytokine involved in inflammatory and immune diseases, such as septic shock (Bacher et al. 1997; Calandra et al. 1995; Bernhagen et al. 1993), glomerulonephritis (Lan et al. 1996,1997,1998,2000), arthritis (Leech et al. 1999), delayed-type hypersensitivity reaction (Bloom and Bennett 1966), atherosclerosis (Lin et al. 2000), and gastric inflammation (Huang et al. 2001). Furthermore, increased serum MIF level has been shown to correlate with the onset of AMI in patients (Yu et al. 2001a). A recent study also observed that plasma MIF level remain elevated 2 weeks after AMI and, importantly its production from the peripheral blood mononuclear cells was increased more than twofold during the same period (Takahashi et al. 2002). In cultured rat cardiac myocytes, significant induction of MIF protein was observed after hypoxia, especially for a more prolonged period (Takahashi et al. 2001). MIF, which is released rapidly from pre-formed intracellular stores, may well be one of the first secreted products to be detected as a consequence of macrophage activation (Calandra et al. 1995). Once released, MIF can stimulate production of tumor necrosis factor-α, inducible nitric oxide synthase, and cellular activation (Calandra et al. 1994,1995). This central regulatory feature of the action of MIF is likely to account for the observation that in vivo MIF immunoneutralization inhibits the expression of various inflammatory mediators in a wide variety of disease models, such as renal disease, gastric ulcer, septic shock, and atherosclerosis (Lan et al. 1997; Calandra et al. 2000; Lin et al. 2000; Huang et al. 2001). A similar mechanism may account for the observation in this study that early and rapid upregulation and secretion of MIF may both initiate and perpetuate the inflammatory response during AMI.
In conclusion, MIF is rapidly and markedly upregulated in myocytes during AMI. Upregulation of MIF by myocytes contributes significantly to macrophage accumulation, and further expression of MIF by infiltrating macrophages may perpetuate the inflammatory response, which may augment the extent of ischemia-induced myocyte damage.
Footnotes
Acknowledgements
Supported by a research grant from CRCG, University of Hong Kong, Hong Kong (HKU7325/99M).