
Abstract
Intracellular signaling mediated by phosphatidylinositol 3-kinase (PI3K) is important for a number of cellular processes and is stimulated by a variety of hormones, including insulin and leptin. A histochemical method for assessment of PI3K signaling would be an important advance in identifying specific cells in histologically complex organs that are regulated by growth factors and peptide hormones. However, current methods for detecting PI3K activity require either homogenization of the tissue or cells or the ability to transfect probes that bind to phosphatidylinositol 3,4,5 trisphosphate (PIP3), the reaction product of PI3K catalysis. Here we report the validation of an immunocytochemical method to detect changes in PI3K activity, using a recently developed monoclonal antibody to PIP3, in paraformaldehyde-fixed bovine aortic endothelial cells (BAECs) in culture and in hepatocytes of intact rat liver. Treatment with either insulin or leptin increased BAEC PIP3 immunoreactivity, and these effects were blocked by pretreatment with PI3K inhibitors. Furthermore, infusion of insulin into the hepatic portal vein of fasted rats caused an increase of PIP3 immunostaining in hepatocytes that was associated with increased serine phosphorylation of the downstream signaling molecule protein kinase B/Akt (PKB/Akt). We conclude that immunocytochemical PIP3 staining can detect changes in PI3K activation induced by insulin and leptin in cell culture and intact liver.
P
Methods currently available for assessing the activity of PI3K require generating cell or tissue homogenates from which biochemical measurements of enzyme activity are made by incubation with radio-labeled substrate (Myers et al. 1992) or by introducing, via transfection, phosphatidylinositol 3,4,5 trisphosphate (PIP3) probes consisting of a PIP3 binding pleckstrin homology domain fused to the green fluorescent protein (GFP) (Varnai et al. 1999; Marshall et al. 2001). These approaches are impractical for determination of PI3K activity in specific cell types of anatomically complex organs such as the brain, which are comprised of heterogeneous cell populations. Therefore, a histochemical method for detecting changes of PI3K activity would be a powerful tool for investigating PI3K signaling in specific cell types that are activated by insulin and leptin in these tissues. Here we report an assay based on the immunocytochemical detection of the principal phosphatidylinositol reaction product of PI3K catalysis to measure relative PI3K activity.
PI3K catalyzes the phosphorylation of phosphatidylinositol (PI) at the 3′-OH position to generate PI(3)P, PI(3,4)P2, and PI(3,4,5)P3 (abbreviated here as PIP3). Physiologically, PI(4,5)P2 appears to be the major substrate for PI3K, with PIP3 being the major reaction product (Katso et al. 2001). Recently, Chen et al. (2002) described the production and initial characterization of a monoclonal antibody (MAb) to PIP3. To generate an immunologically active hapten, they developed acyl-tethered PIP3 compounds for use as immunogens and succeeded in generating an MAb (RC6F8) that has 1000-fold selectivity for PIP3 over PIP2 in binding assays. Finally, the authors showed that this antibody is capable of immunohistochemically detecting PIP3 in NIH-3T3 cells stimulated by insulin and platelet-derived growth factor, as well as in stimulated human neutrophils (Chen et al. 2002).
Here we report the use of this PIP3 antibody to detect increased PIP3-like immunostaining of primary bovine aortic endothelial cells (BAECs) that were stimulated in vitro by insulin and leptin. Using a semiquantitative approach, immunological controls, and PI3K inhibitors, we demonstrate that the PIP3-like immunoreactivity detected by this antibody arises from activation of PI3K. Insulin and leptin stimulation resulted in a marked enhancement of immunoreactive PIP3 over a timecourse consistent with the well-characterized biochemical actions of these hormones (Zeng and Quon 1996; Kim et al. 2001). We also show that immunoreactive PIP3 co-localizes with the insulin signal transduction pathway molecule insulin receptor substrate-2 (IRS-2). Finally, we report that this method is applicable to an in vivo system in which enhanced PIP3 immunofluorescence is detected in rat liver after insulin stimulation and corresponds with activation of protein kinase B/Akt (PKB/Akt), a downstream target of PI3K signaling.
Materials and Methods
Cell Culture Studies
Cultured BAECs were established and maintained in DMEM supplemented with 10% fetal bovine serum, glutamine, and penicillin/streptomycin (Corson et al. 1996) on gelatin-coated glass microscope coverslips in 8-well plates. After being serum-starved for 2.5 hr in endothelial cell basal medium (Clonetics; Walkersville, MD), cells were pretreated with an inhibitor of PI3K: either 20 μM LY294002 (Calbiochem; San Diego, CA) or 100 nM wortmannin (Calbiochem) for 10 min. Controls received only the vehicle for 10 min. Cells were then incubated with either 100 nM insulin (Sigma; St Louis, MO) or 100 nM leptin (Peprotech; Rocky Hill, NJ) for 5 min, after which the cells were washed twice in PBS, fixed for 5–10 min in 4% paraformaldehyde in 0.1 M phosphate buffer (Electron Microscopy Sciences; Ft Washington, PA), and washed three times in PBS.
Studies In Vivo
All procedures involving the use of animals were approved by the Animal Research Committees at the University of Washington and the VA Puget Sound Health Care System. Mature (300–340 g) male Wistar rats (Simonsen; Gilroy, CA) were housed under standard conditions. Animals were fasted for 48 hr (with water freely available), then anesthetized with a ketamine/xylazine mixture. A 5-mU injection of insulin in saline (n = 3) (or saline alone, n = 3) was administered via the hepatic portal vein, followed 15 min later by portal vein perfusion with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). Liver samples were removed and placed in fresh fixative overnight, followed by the the buffer alone containing 25% sucrose (w/v) overnight at 4C. They were snap-frozen in freon cooled by dry ice. Cryostat sections (14 μm) were mounted on glass slides and stored at −70C.
Immunohistochemical Methods
Coverslips containing BAECs or slides with liver sections were equilibrated in PBS at room temperature. After blocking in 5% normal goat serum (NGS) and 2% bovine serum albumin (BSA), samples were incubated with mouse anti-PIP3 monoclonal antibody (Echelon; Salt Lake City, UT) at a 1:100 dilution overnight at 4C. Dilutions ranging from 1:50 to 1:500 were tested, with 1:100 giving the best signal-to-noise ratio. The negative immune control for this antibody was an equivalent concentration of non-immune mouse IgM (DakoCytomation; Carpinteria, CA) substituted for the primary antibody at an equivalent dilution. All antibodies were diluted in 0.1 M PBS containing 2% BSA and 5% NGS. All BAEC samples were counterstained with DAPI (Molecular Probes; Eugene OR) to count the number of nuclei per field of view. To detect PI3K-mediated serine phosphorylation of PKB/Akt- as a measure of activation of the PI3K pathway, we used a polyclonal antibody (1:100) to serine473-phosphorylated PKB/Akt (Cell Signaling Technology; Beverly, MA). The IRS-2 antibody [gift of M. Myers Jr, Joslin Diabetes Center, Harvard Medical School, Boston, MA (Lingohr et al. 2002)] was used at a dilution of 1:300. Primary antibodies were detected with either Alexa488 (Molecular Probes) or Cy3 (Jackson Immunoresearch; West Grove PA)-conjugated goat anti-mouse or goat anti-rabbit antibodies at a dilution of 1:200. Sections/cells were mounted in aqueous polyvinylalcohol mounting medium.
Imaging Methods
Images of BAECs were captured via standard epifluorescence using a Nikon Eclipse E600 upright microscope equipped with a Diagnostic Instruments Spot RT Color digital camera. All images for a single experiment were obtained using identical acquisition parameters in one imaging session. PIP3 immunofluorescence was quantified using Scion Image freeware (Scion; Frederick, MD) as the simple average pixel intensity for the field of view. DAPI fluorescence was evaluated via particle analysis using Scion Image freeware, yielding the total number of cells present per field of view. Values are reported as the mean± SEM of 6–10 fields of view per coverslip and at least two to four coverslips per measurement. The average pixel intensity was analyzed in this way from a set of coverslips (n = 3) in which an equivalent concentration of non-immune mouse IgM was substituted for the PIP3 antibody. The average pixel intensity derived from these samples, representing cell autofluorescence and nonspecific immune interactions, was subtracted from the average pixel intensity derived from the experiments described in Figures 2 and 3 to report a “corrected” cell fluorescence that more accurately reflects the true increase in immunodetectable PIP3 on hormone stimulation. Statistical significance was determined by ANOVA with Bonferroni's post test for multiple comparisions. Although separate experiments were not combined because we could not ensure identical staining and imaging parameters during different sessions on different days, the results are consistent among different experiments. Images of rat liver were acquired with a Zeiss Axiophot fluorescence microscope.
Results
PIP3 Is an Immunodetectable Molecular Species in Cells
PIP3 immunoreactivity was readily visible in serum-fed BAECs stained with anti-PIP3 antibody (Figures 1A and 1B) but was not detected in cells incubated with an equal concentration of non-immune mouse IgM (Figures 1C and 1D). In comparison to serum-starved BAECs, PIP3 immunoreactivity was visibly increased in serum-fed cells and in cells stimulated by insulin or leptin (Figure 2).
Insulin and Leptin Stimulate PIP3-like Immunoreactivity in BAECs
Cultured BAECs are responsive to both leptin (Artwohl et al. 2002) and insulin (Zeng and Quon 1996; Kim et al. 2001), and both hormones are known to signal through PI3K (Kellerer et al. 1997; Saltiel and Pessin 2002). To determine the effect of insulin and leptin on PIP3 immunofluorescence in a semiquantitative fashion, BAECs were serum-starved for 2.5 hr and then treated with either vehicle, insulin or leptin, and PIP3 fluorescence quantitated by pixel intensity. Insulin (Figures 2B and 2D) and leptin (Figures 2C and 2D) increased PIP3 immunofluorescence by 2.7± 0.1 and 3.6± 0.2-fold, respectively (p<0.05 for each; n = 16–24 fields over two to three coverslips) relative to serum-starved, vehicle-treated cells (Figures 2A and 2D). To rule out a mitogenic effect of either hormone or a random increase in the number of cells per field of view accounting for higher PIP3 signal, a corresponding DAPI nuclear stain image was collected for each PIP3 fluorescence image obtained. There was no difference in the average number of cells per field of view for vehicle (80± 4) vs insulin (85± 3) or leptin (77± 3; p = nonsignificant among the groups).
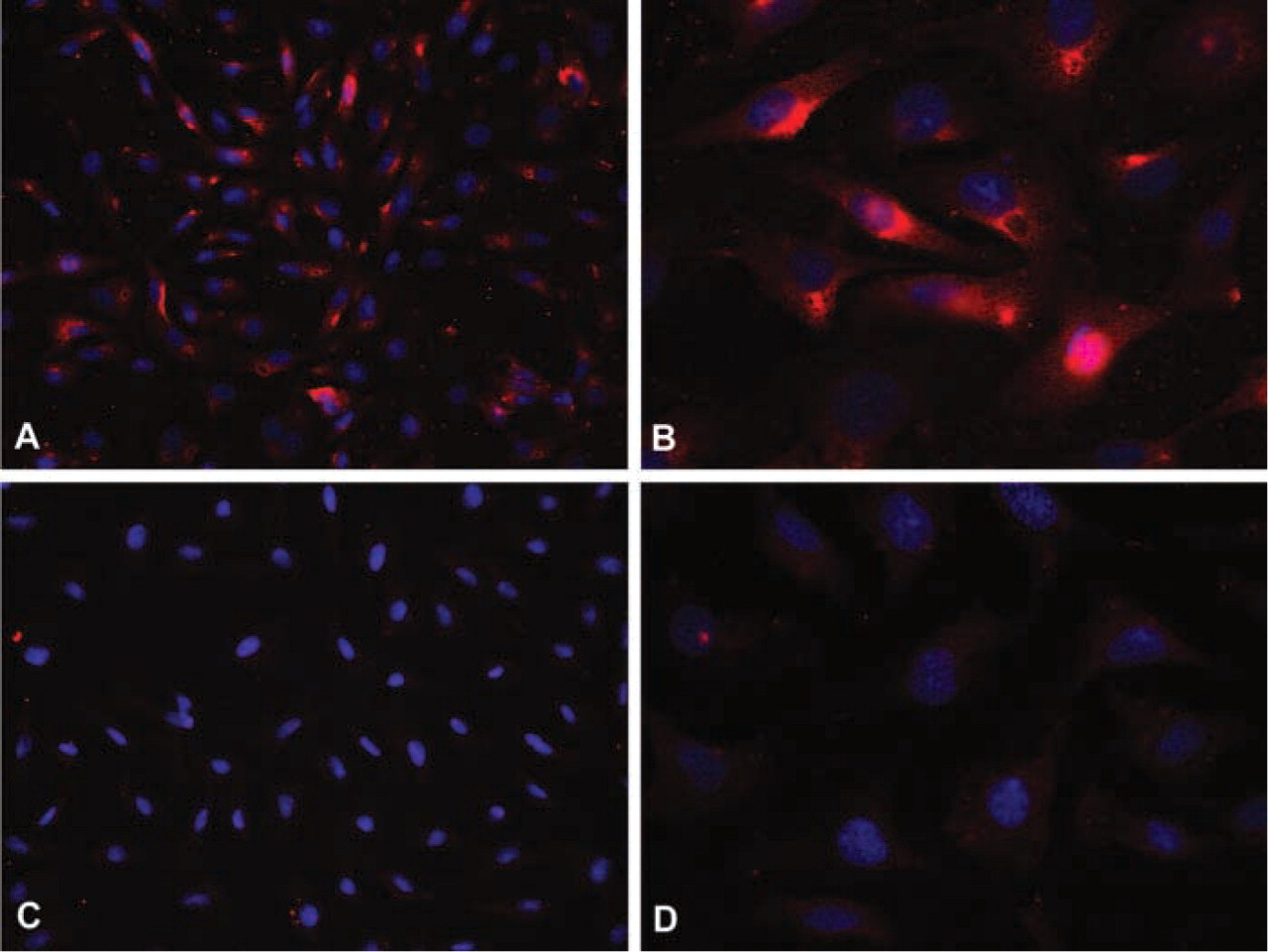
PIP3 is an immunodetectable species. BAECs cultured under standard conditions with serum demonstrate significant cytoplasmic and membrane staining for PIP3 at both × 40 (
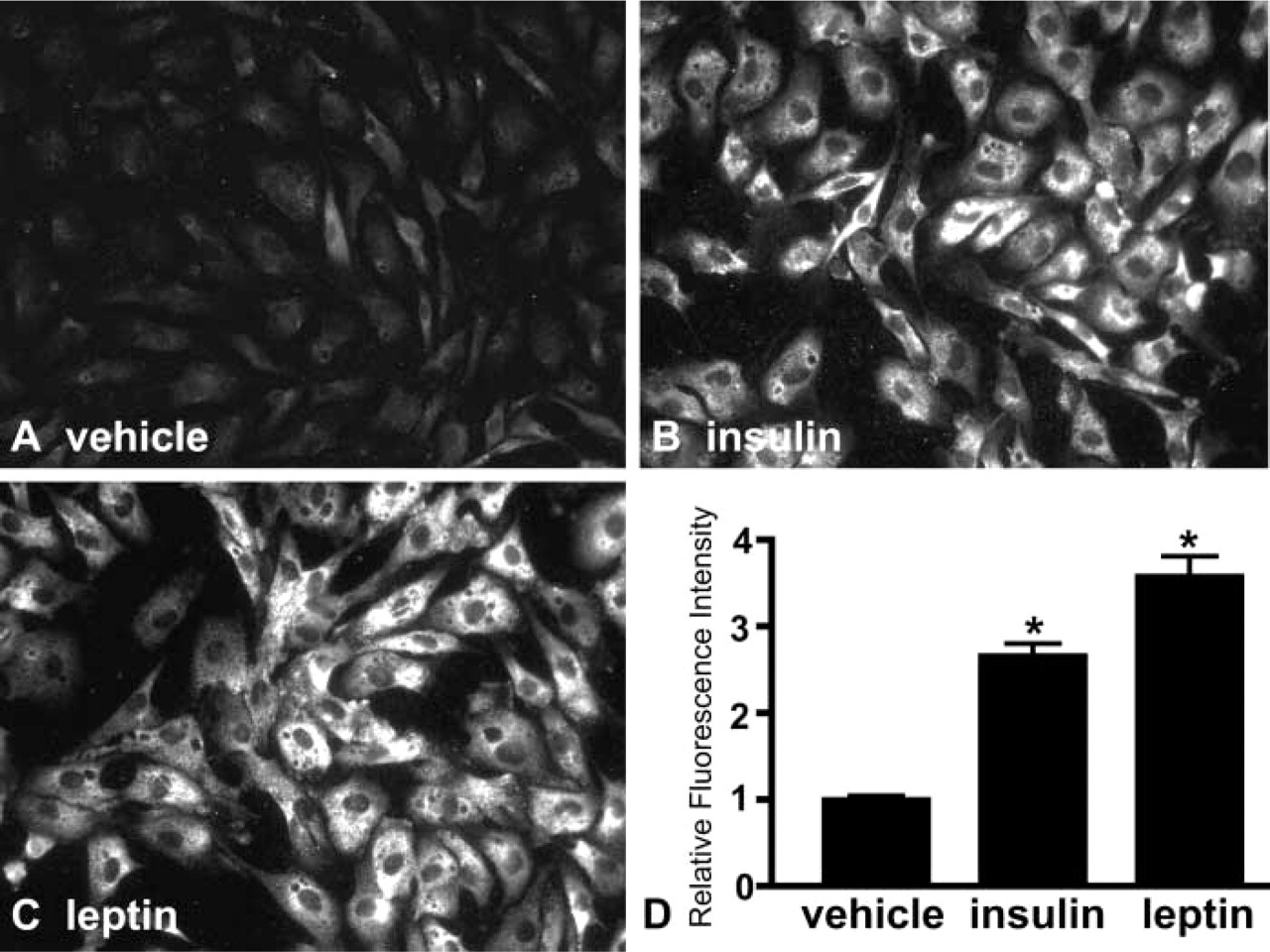
Insulin and leptin induce PIP3 immunoreactivity in BAECs. BAECs were serum-starved for 2 hr, then treated with insulin (
Immunoreactive PIP3 Arises from Activation of PI3K
To ensure that increased PIP3 immunofluoresence in BAECs treated with leptin was due specifically to activation of PI3K, rather than to a nonspecific effect, cells were pretreated with either of two PI3K inhibitors, LY294002 (Figures 3C and 3E) or wortmannin (Figures 3D and 3E) or with vehicle, followed by a 10-min incubation with leptin. Pretreatment with either PI3K inhibitor before hormone treatment blocked the increase of PIP3 immunoreactivity (Figures 3A and 3E) observed when cells were pretreated with vehicle followed by leptin (Figures 3B and 3E). Similar findings were observed with insulin treatment (data not shown). Because PI3K is the only enzyme known to be inhibited by both wortmannin and LY294002, we conclude that leptin-induced increases of PIP3 immunofluoresence in BAECs resulted specifically from activation of PI3K.
Immunoreactive PIP3 Co-localizes with IRS-2
To determine whether the PIP3-like immunoreactivity observed after insulin stimulation could represent PIP3 that participates in intracellular signaling, we sought to determine whether the insulin receptor-PI3K coupling molecule IRS-2 co-localizes with PIP3 on stimulation by insulin. In the vehicle-treated state there is relatively weak immunocytochemical co-localization of IRS-2 (green fluorescence) with PIP3 (red fluorescence) (Figure 4A), indicated by the relative absence of yellow (red plus green) fluorescence. However, 5 min of insulin treatment resulted in a substantially increased immunological co-localization of IRS-2 and PIP3 (Figure 4B). The areas of greatest co-localization included the perinuclear area and the plasma membrane, where intracellular targets of PI3K activation are known to exist (Sessa et al. 1995; Varnai et al. 1999). Replacement of the IRS-2 serum with an equivalent amount of non-immune serum resulted in no visible signal under the same acquisition parameters (not shown). This finding is consistent with the hypothesis that increased intracellular levels of PIP3 induced by insulin result in a close physical association with signaling molecules such as IRS-2, either via an interaction of PIP3 with the PIP3 binding domain of IRS-2 (pleckstrin homology domain) or via the interaction of the regulatory subunit of PI3K with IRS-2 or the insulin receptor per se. In either case, this finding implies that PIP3 induced by insulin treatment and detected by this antibody participates in intracellular signal transduction, given its close physical association with the signaling apparatus.
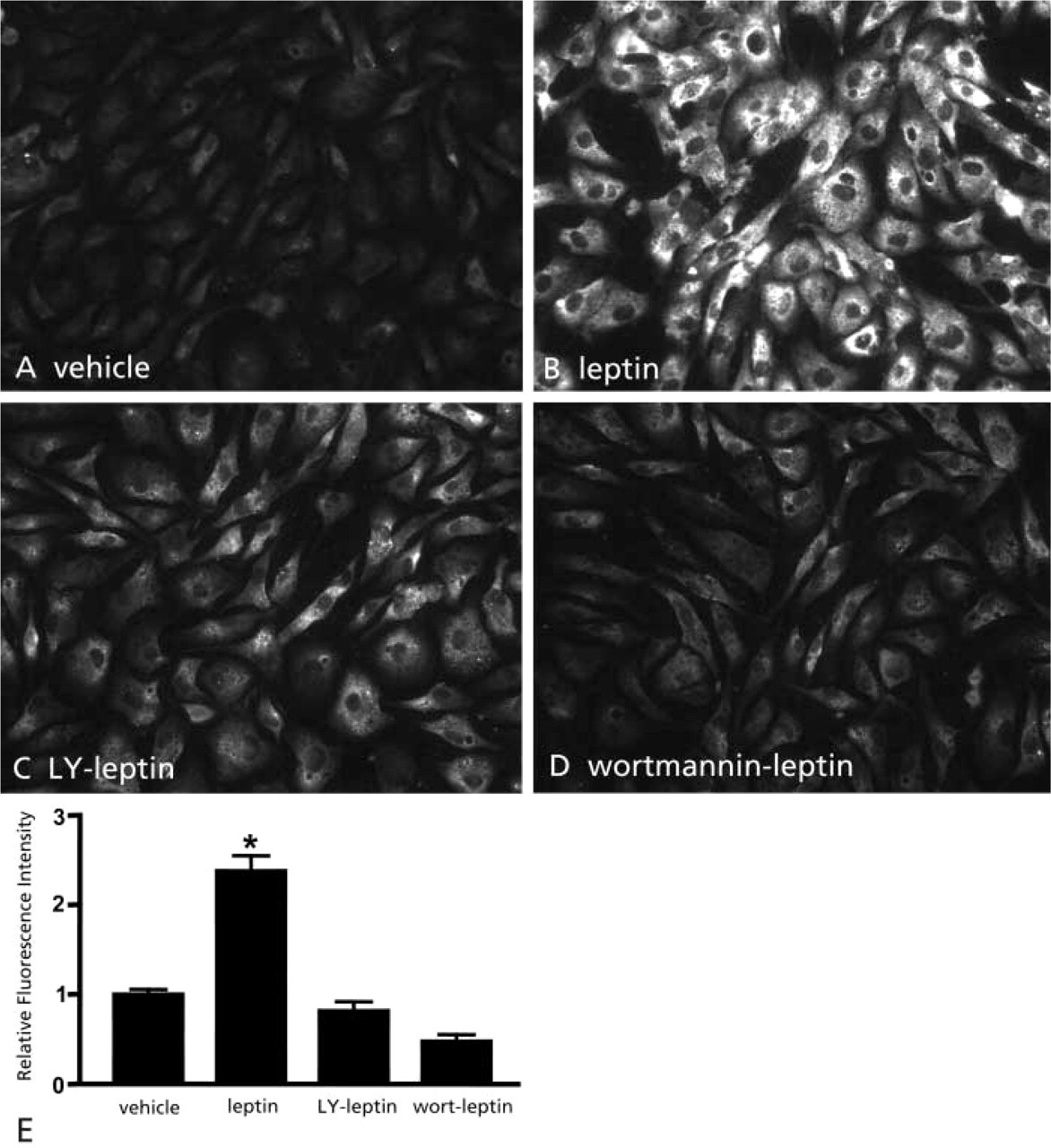
Leptin induces PIP3 immunoreactivity in BAECs by activating PI3K. Leptin (
Insulin Stimulation Generates Immunoreactive PIP3 and Phosphorylated PKB/Akt in Hepatocytes In Vivo
To determine whether the PIP3 immunoreactive species detected by this antibody could be used to detect changes in PI3K activity in situ, we examined PIP3 immunofluorescence in liver obtained from rats infused with either insulin or saline directly into the hepatic portal vein 15 min before sacrifice, a protocol that induces activation of insulin receptor signaling and phosphorylation of IRS proteins and PI3K in rat liver (Saad et al. 1993; Carvalho et al. 1996). Insulin induced a clear increase in hepatocyte PIP3 immunofluorescence (Figure 5D vs 5C), consistent with activation of PI3K by insulin. Importantly, PIP3 fluorescence was enhanced in the zone of cells surrounding the portal vein that would be expected to be exposed to the highest concentrations of insulin via the hepatic portal vein perfusion. Enhancement of PIP3 immunoreactivity was mirrored by increased serine473 phosphorylation of PKB/Akt (Figure 5B vs 5A), a downstream target of PI3K/PIP3 activation, which is consistent with the conclusion that the PIP3 antibody revealed changes in PI3K activity in the insulin-stimulated hepatocytes.
Discussion
Our objective in developing this immunocytochemical method for detecting insulin- and leptin-stimulated PIP3 was to validate a direct assay of the activation of PI3K in histologically intact tissue. Current methods of quantifying PI3K activity require homogenization of the tissue or cells of interest, followed by biochemical analysis. Such methods work well for reasonably homogeneous tissues, such as liver and skeletal muscle, and for experiments in which information on cell variability, cell phenotype, or the response of individual cells is not required. They are obviously not appropriate for anatomically complex heterogeneous tissues, such as brain, in which the cell type of interest may be quite rare. The generation of an antibody that specifically recognizes PIP3 therefore promises to have great value in morphological studies of PI3K signaling. Chen et al. (2002) and Chellaiah et al. (2001) have demonstrated specificity of the antibody for PIP3 using a series of biochemical assays, and a “lipid” blot. However, inositol phospholipids may be ubiquitous in cells and are a key constituent of cell membranes. Biochemical specificity therefore does not ensure that the antibody will perform as predicted in immunohistochemical assays for the purposes of examining cell signaling. Therefore, we sought to extend the findings of Chen et al. (2002) by performing immunological controls and by studying the utility of the antibody in a well-characterized system known to utilize PI3K signaling.
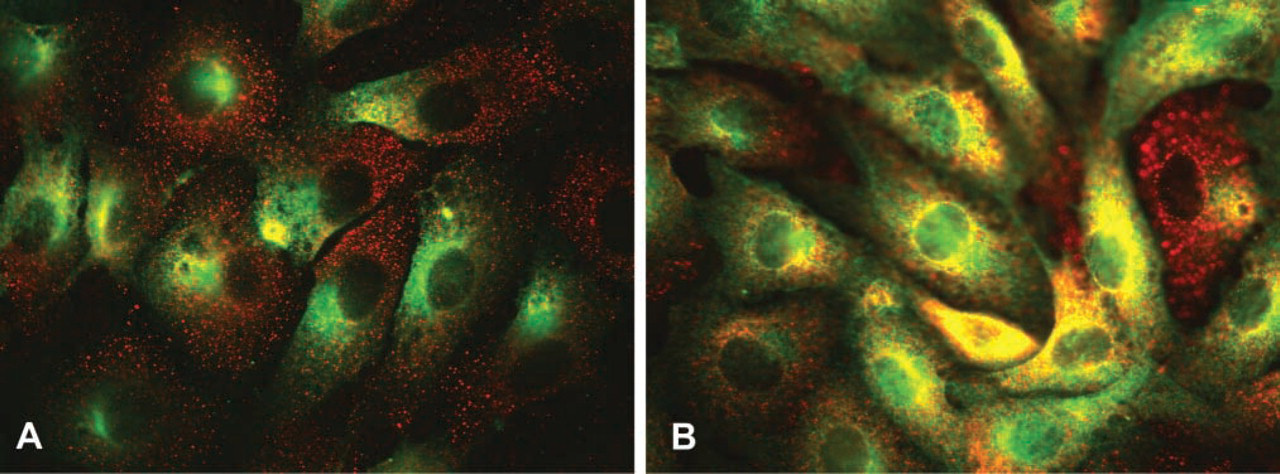
PIP3 co-localizes with insulin receptor substrate 2 (IRS-2) on insulin treatment. Serum-starved BAECs in the absence of insulin treatment (
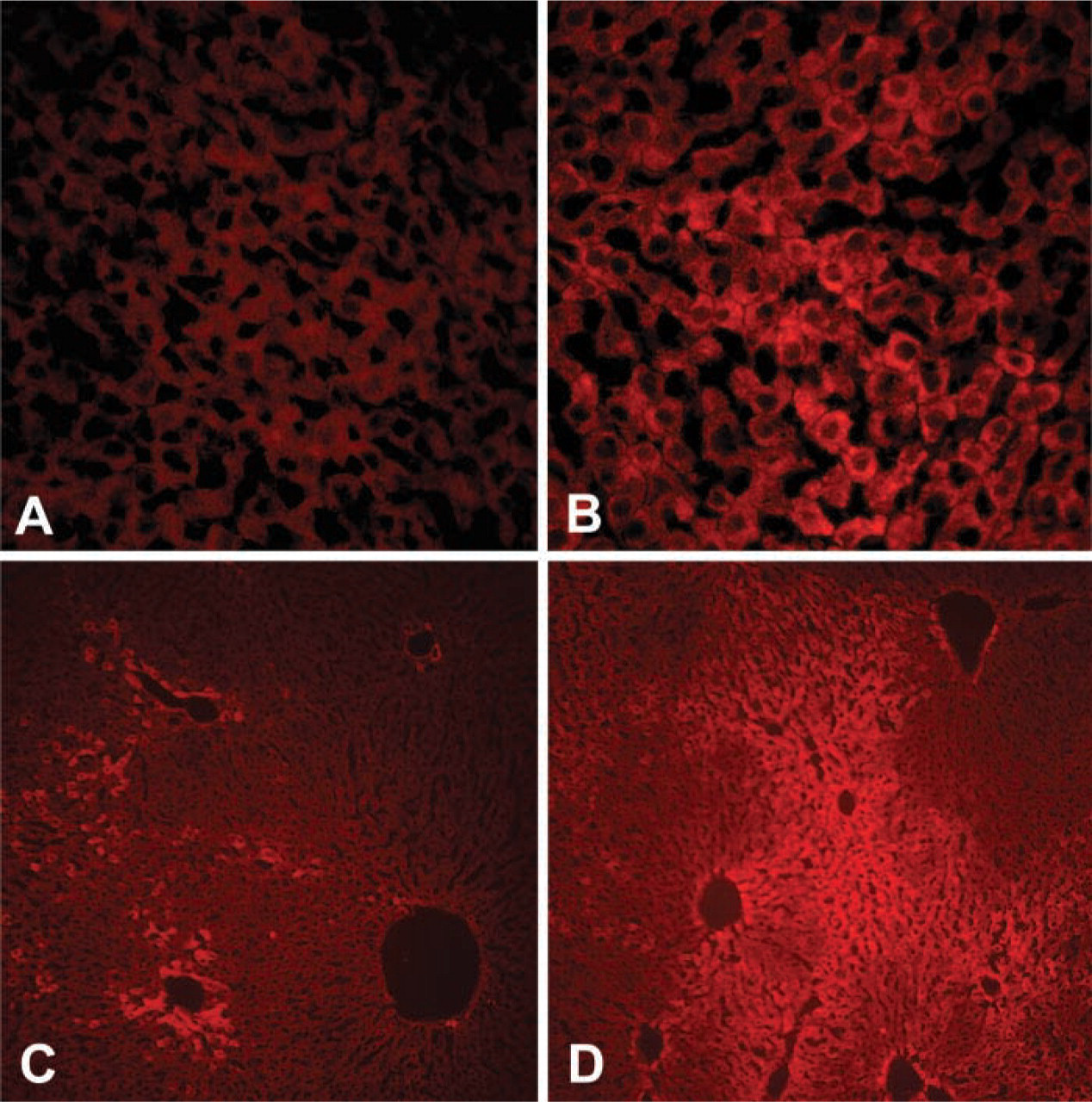
Insulin induces PIP3 fluorescence and activation of PKB/Akt in in situ perfused rat liver. Compared with vehicle infusion (
The signaling cascade leading to activation of PI3K and generation of PIP3 is initiated by growth factor or hormone binding to its respective receptor, activating either an intrinsic tyrosine kinase (insulin) or an extrinsic tyrosine kinase (janus kinase in the case of leptin). Phosphorylation of the intracellular portion of the majority of these receptors by their respective kinase results in recruitment and binding of downstream signaling molecules to the phosphorylated tyrosine residues. In the case of insulin, receptor activation in this manner results in the tyrosine phosphorylation and activation of one of a family of insulin receptor substrate molecules (IRS 1–4) which, in turn, binds to the regulatory subunit of PI3K (Saltiel and Pessin 2002). Similarly, leptin binding to and activation of its receptor results in the activation of janus kinase (JAK), which has also been suggested to phophorylate IRS molecules and activate PI3K (Anderwald et al. 2002). Binding of the regulatory domain of PI3K serves both to relieve inhibition of the PI3K catalytic subunit and to recruit the enzyme complex to membranes in which its substrate [PI(4,5)P2] is located. PIP3 produced at the membrane in turn recruits and activates a number of other downstream molecules that contain PIP3 binding domains (Rameh et al. 1997), including serine-threonine kinases, tyrosine kinases, GTPases, and others. For example, in signaling mediated by insulin activation of PI3K, downstream molecules that are activated include PDK1, PKB/Akt and, in endothelial cells, eNOS (Gallis et al. 1999; Kim et al. 2001; Saltiel and Pessin 2002).
An emerging area of great interest in the fields of diabetes and obesity research is the concept of “crosstalk” or “convergence” between insulin and leptin intracellular signaling pathways. In several cell types, including muscle cells (Kellerer et al. 1997), adipocytes (Kim et al. 2000), β-cells (Harvey et al. 2000), and hypothalamic neurons, both insulin and leptin activate PI3K, which may account, at least in part, for similarities in the metabolic effects of these hormones (Niswender and Schwartz in press). A method for the detection of PI3K activation in situ, such as described here, provides a new tool to investigate the cell, tissue, and physiological ramifications of such crosstalk.
The experiments reported here demonstrate that immunocytochemistry with an antibody to PIP3 reveals increased immunofluorescence of BAECs treated with insulin and leptin and that this effect was blocked with PI3K inhibitors. These observations are consistent with PI3K catalysis stimulated by insulin and leptin. Although neither PI3K inhibitor is entirely specific, the only enzyme known to be inhibited by both LY294002 and wortmannin is PI3K (Davies et al. 2000). Given that both PI3K inhibitors block the induction of PIP3-like immunofluorescence and that both are known to block the activation of downstream targets of PI3K in BAECs (Gallis et al. 1999), we conclude that the increase of PIP3 immunofluorescence observed in the absence of PI3K inhibitor resulted primarily from activation of PI3K. This critical experiment strongly supports the use of this PIP3 antibody as a tool for histochemical analysis of PI3K signaling.
Interestingly, the PIP3-like immunofluorescence levels in unstimulated cells were quite low, as expected, and there was very little co-localization with IRS-2. On insulin stimulation, marked enhancement of co-localization with IRS-2 was observed, particularly in the region occupied by perinuclear membranes and in the vicinity of the plasma membrane, both sites at which targets of PI3K signaling are known to exist (Sessa et al. 1995; Saltiel and Pessin 2002). Therefore, hormone treatment results in an intracellular pattern of spatial association of PIP3 and IRS-2 that is consistent with the accepted location of this intracellular signaling pathway (Saltiel and Pessin 2002). Although other interpretations are possible, we conclude that the increases in PIP3-like immunofluorescence that were induced by hormone treatment in the present study represent PIP3 that actively participates in intra-cellular signal transduction. More direct evidence for a signaling role of insulin-induced PIP3-like immunore-activity comes from staining in vivo insulin-treated hepatocytes in which increased PIP3-like immuno-staining was associated with elevated immunostaining for serine473-phosphorylated PKB/Akt, a signaling molecule downstream from PI3K/PIP3 activation. These findings are all consistent with the hypothesis that, under appropriate experimental conditions, PIP3-like immunocytochemistry can reveal changes in PI3K activity induced by insulin and leptin. In addition, compared to the saline vehicle control, insulin induced a visible increase in PIP3-like immunofluorescence primarily in the regions surrounding the hepatic portal veins, a physiologically appropriate histological distribution. Therefore, in addition to cell culture, we anticipate success in using this methodology in a variety of heterogeneous tissues in which specific cells utilize PI3K signaling.
Because PI3K is an evolutionarily conserved intra-cellular signaling system (Wolkow et al. 2000; Garofalo 2002) that is important for a variety of cellular processes (Katso et al. 2001), a histochemical method for detecting changes in PI3K activity provides an approach to identifying PI3K activation in specific cell types in situ. The present investigation suggests that immunocytochemistry of PIP3, the principal PI3K catalysis product, using the PIP3 antibody recently developed by Chen et al. (2002) is a valid and useful tool for detection of changes in intracellular PI3K signaling.
Footnotes
Acknowledgements
Supported by NIH grants DK17047 to the Diabetes Endocrinology Research Center at the University of Washington, Department of Veterans Affairs Career Scientist and Merit Review Research Programs to DGB, and NIH grants NS32273, DK12829, and DK52989 to MWS, and HL30946 to MAC. KN is supported by NIH Training Grant T32 DK07247.
We acknowledge the skilled technical efforts of J. Murphy and T. Eakin.