
Abstract
Interconnection between surface microdomains and the actin cytoskeleton is vital to various cellular activities. We studied the responses of okadaic acid (OKA)-treated K562 leukemia cells to crosslinking of membrane microdomains. Although OKA alone induced clustering of surface-bound F-actin, addition of a biotinylated poly(ethylene glycol) derivative of cholesterol (bPEG-Chol) and subsequent binding of streptavidin (SA) further induced accumulation of the clusters, resulting in the formation of a spherical cell extrusion. This extrusion was also induced by direct crosslinking of a raft marker, CD59, and ganglioside GM1. In addition, we found that knockout of the gene encoding Fyn kinase inhibited formation of the spherical extrusion in murine T-cells. In bPEG-Chol/SA-treated cells, CD59, ganglioside GM1, and clathrin/AP-2 were all accumulated on the surface of the actin-rich extrusion, whereas dynamin and transferrin receptors were unaffected. Intermediate filaments, mitochondria, and other vesicles also accumulated. These results suggest that crosslinking of membrane domains exaggerates the linkage between actin and a defined set of membrane proteins in OKA-treated cells.
L
Distinct sets of proteins that regulate endocytic pits also control cytoskeleton-dependent cell activities (for review see Geli and Riezman 1998; Qualmann et al. 2000). Dynamin and a dynamin-interacting protein, amphiphysin, control cell motility and shape (Damke et al. 1994; Mundigl et al. 1998). Among the various SH3-containing proteins that regulate budding of coated pits at distinctive steps, syndaptin interacts with dynamin and N-WASP-Arp2/3 (Qualmann and Kelly 2000). An actin-binding protein, Abp1 (SH3P7/HIP-55), binds to dynamin and is enriched in endocytosis-active membrane regions (Kessels et al. 2001). Another actin-binding protein, Sla2 (HiP1R; Huntingtin-interacting protein-1 related), is enriched in clathrin-coated pits and vesicles (Engqvist-Goldstein et al. 1999). Actin is crucial for endocytosis itself, because depolymerization of F-actin by latrunculin B interferes with the distribution (Gaidarov et al. 1999) and closure (Lamaze et al. 1997) of clathrin-coated pits. In contrast, hyperstabilization of microfilaments by jasplakinolide inhibits endocytosis of fluid markers (Shurety et al. 1998).
In addition to these cytoskeletal systems, protein phosphorylation is crucially involved in the regulation of endocytosis and cell motility. In particular, okadaic acid (OKA), an inhibitor of ser/thr protein phosphatases 1 and 2A, induces cell rounding and inhibits many endocytic steps (Lucocq et al. 1991; Mundy et al. 1991; Woodman et al. 1992) and cell motility (Wilson et al. 1991; Downey et al. 1993; Draskovic-Pavlovic et al. 1999). When adherent cells are treated with OKA, caveolae are internalized in an actin-dependent manner (Parton et al. 1994). Functional and structural study of the linkage between actin and lipid rafts/clathrin-coated pits using OKA may help to elucidate the normal mechanism but has not been addressed systematically.
Here we describe the linkage of actin to lipid rafts and clathrin-coated pits in OKA-treated K562 leukemia cells. We addressed different strategies that are expected to aggregate surface domains or to deform the plasma membrane and cortex actin. One of the methods involved distributing a biotinylated poly(ethylene glycol) derivative of cholesterol (bPEG-Chol; see Figure 1), which was subsequently crosslinked by streptavidin (SA). The major advantage of this amphiphilic molecule is that an inserted cholesteryl moiety does not interfere with the lateral interactions between adjacent lipids. This is supported by previous results that PEG-Chol-containing liposomes effectively encapsulated water-soluble drugs without disturbing membrane structures (Ishiwata et al. 1995,1998). We have recently found that bPEG-Chol is preferentially inserted in lipid rafts (unpublished results). We also crosslinked lipid rafts by a combination of antibodies. To deform the plasma membrane by physical force, we addressed the effect of pressing the cells via a coverslip. Remarkably, all these methods induced accumulation of actin in a single pole. Using this method, we compared the distribution of raft- and clathrin-coated pit-associated elements to that of actin. We suggest that various membrane stresses on OKA-treated cells exaggerate and also differentiate the linkage between cortical actin with defined sets of membrane components.
Materials and Methods
Cells
K562 cells were maintained in DMEM/10% FCS. The cells were incubated in serum-free medium (SFM, DMEM supplemented with 0.2% BSA and 20 mM Hepes, pH 7.4) for 30 min before the experiment. The cells were pelleted and suspended in SFM at a concentration of 5 × 106 cells/ml.
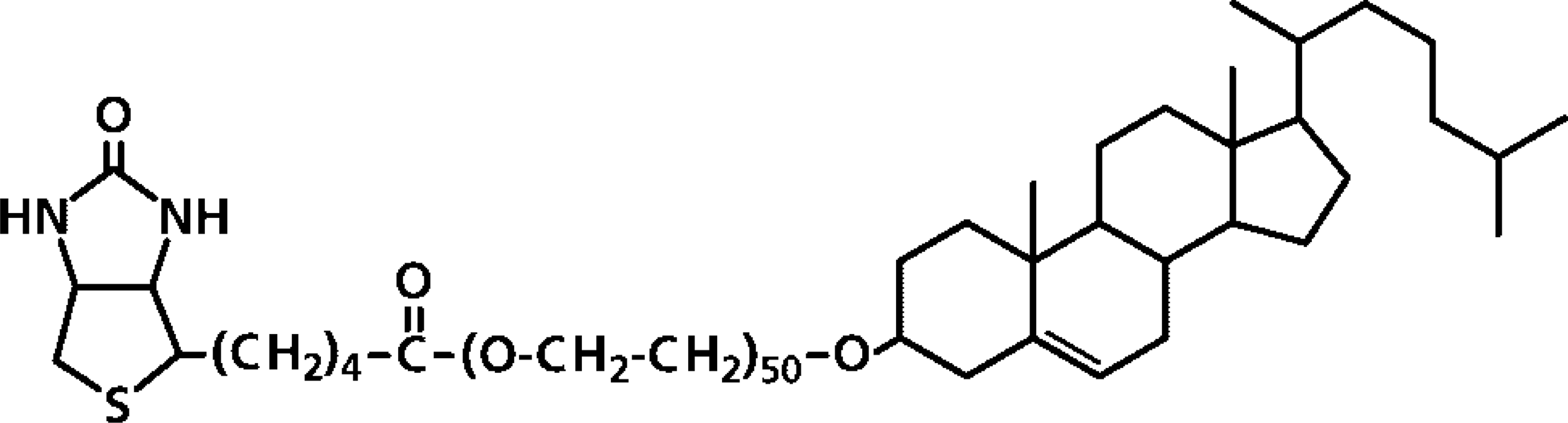
Structure of biotinylated poly(ethylene glycol) cholesteryl ether (bPEG-Chol).
Reagents
Poly(ethylene glycol)50-cholesteryl ether (PEG-Chol) and biotinylated PEG-Chol (bPEG-Chol) were prepared as previously reported (Ishiwata et al. 1997; Baba et al. 2001). Okadaic acid (OKA) was purchased from Wako Pure Chemical (Tokyo, Japan). Jasplakinolide, latrunculin A, MitoTracker Red CMXRos, AlexaFluor594-streptavidin, Alexa Fluor488-phalloidin, and Alexa Fluor488-goat anti-mouse IgG were purchased from Molecular Probes (Eugene, OR). Anti-CD45 and anti-CD59 MAbs were purchased from Pharmingen (San Diego, CA). Anti-integrin β1 antibodies, TS2/16 (Endogen; Woburn, MA), and DF5 (Chemicon; Temecula, CA) were purchased from the indicated distributors, respectively. All other chemicals were purchased from Sigma (St Louis, MO).
Transfection of pEGFP-Actin in K562 Cells
A plasmid containing a gene encoding pEGFP-human-β-actin was purchased from Clontech (Palo Alto, CA). For transfection, K562 cells were incubated overnight with an iron-depleted medium. The cells were transfected with a Transferrinfection kit (Takara; Kyoto, Japan) according to the manufacturer's protocol. The cells were used for experiments 2 days after transfection.
OKA/b-PEG-Chol/Streptavidin Treatment
K562 cells in SFM were incubated with 500 nM OKA for 30 min at 37C and with 2 μM bPEG-Chol (2 nmol/5 × 106 cells) for another 30 min at 37C without washing out OKA. The cells were chilled on ice and incubated with 10 μg/ml AlexaFluor594–streptavidin (SA) for 30 min at 4C. These cells were then incubated for 5–30 min at 37C and fixed with 4% paraformaldehyde and 0.1% glutaraldehyde in 0.1 M phosphate buffer for 15 min at 4C. The cells were washed with PBS and mounted in PBS/BSA/Glycerol.
Immunocytochemistry
For immunolocalization of cell surface antigens, OKA/bPEG-Chol/SA-treated cells were fixed as above, attached to coverslips by a cytofuge (Statspin; Norwood, MA), and incubated with mouse MAbs for 30 min at RT. The cells were washed in PBS and reacted with AlexaFluor 488-goat anti-mouse IgG for 30 min. For immunolocalization of cytoplasmic antigens, fixed cells were attached to coverslips by the cytofuge and treated with 0.1% saponin/PBS for 10 min at RT. The cells were washed in PBS, blocked with 1% BSA/PBS, and incubated with primary antibodies or with AlexaFluor 488-phalloidin for F-actin. These samples were observed in a confocal laser-scanning microscope (Leica TCS4D; Heidelberg, Germany).
Fyn(−/−) Murine T-cells
Murine fyn(+/+) and fyn(−/−) T-cell lines were established from a 2C TCR transgenic mouse (Sha et al. 1988) and its cross with an fyn-knockout mouse (Yagim et al. 1993), respectively. T-cell lines were stimulated weekly with irradiated P815 cells in 10% FCS DME containing 5% rat ConA culture supernatant. These cells were treated with 500 nM OKA for 30 min, then with bPEG-Chol/SA at 4C for 30 min, and finally at 37C for 30 min. After fixation, the cells were immunostained with anti-Thy-1 MAb (Pharmingen). These cells were observed in an Olympus BX50 microscope (Olympus; Tokyo, Japan) equipped with a Hamamatsu ORCA-2 camera (Hamamatsu Photonics; Hamamatsu, Japan) controlled by imaging software Metaview (Universal Imaging; West Chester, PA).
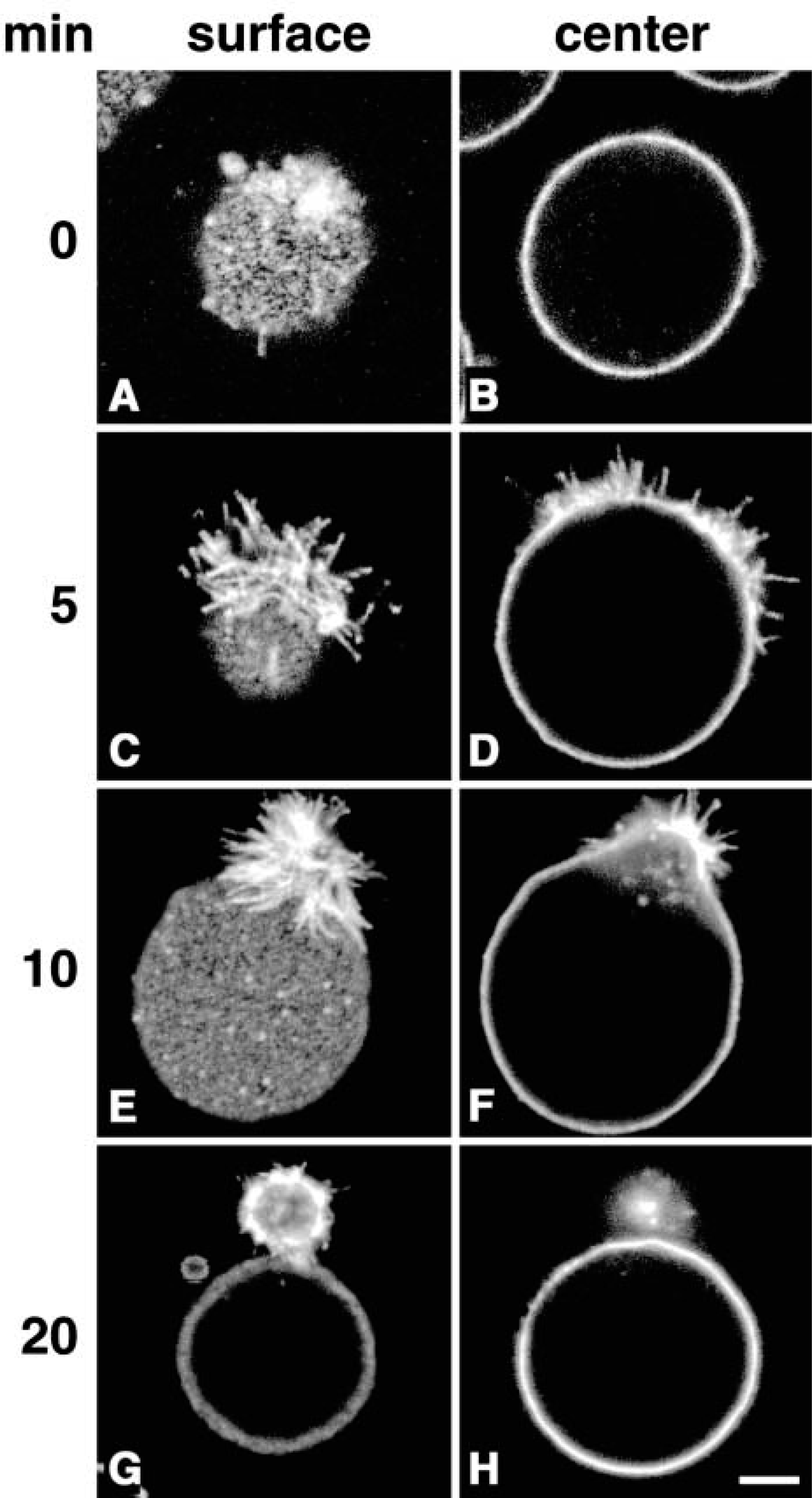
Formation of a spherical extrusion in OKA-treated K562 cells by bPEG-Chol/SA. K562 cells (5 × 106 cells/ml) were treated with 500 nM OKA for 30 min, then with 2 μM bPEG-Chol for 30 min at 37C. They were then chilled in ice and incubated with Alexa-Fluor594-labeled SA for 30 min. After washing, cells were observed in a confocal microscope after 0 min (
Results
K562 cells were treated with okadaic acid (OKA, 500 nM) at 37C for 30 min. The cells were then incubated with bPEG-Chol at 2 nmol to 5 × 106 cells at 37C for 30 min. The structure of bPEG-Chol is shown in Figure 1. Because 2 nmol of PEG-Chol is known to occupy approximately 1% of total plasma membrane area, incorporated bPEG-Chol is equivalent to several percent of total plasma membrane cholesterol (Baba et al. 2001). The incorporated molecule was bound with AlexaFluor 594-SA at 4C. At this temperature, SA distributed homogenously over the surface with slight accumulation (Figures 2A and 2B). When the cells were incubated at 37C, SA accumulated markedly in filopodium-like protrusions (Figures 2C and 2D). With further incubation, the structures became segregated from the main cell body (Figures 2E and 2F, referred to as budding), and finally separated as a single spherical bud (Figures 2G and 2H, referred to as fission). These data are summarized in Figure 3. Similar results were obtained with pretreatment with 100 nM calyculin A, which had a broader spectrum as a phosphatase inhibitor (data not shown). OKA at 100 or 200 nM, which does not inactivate some phosphatases, did not induce these changes (data not shown). No spherical buds were observed in bPEG-Chol/SA-treated cells without OKA treatment (data not shown), as previously reported for PEG-Chol (Baba et al. 2001).
The distribution of F-actin was visualized using fluorescent phalloidin in comparison with the accumulation of bPEG-Chol/SA (Figure 4). At 4C, actin was already clustered on the surface (Figure 4A). When the cells were incubated at 37C, co-migration of both molecules to the spherical extrusion was clearly seen (Figures 4D–4F). A similar change in the pattern of fluorescence was observed in living K562 cells expressing EGFP-actin (Figures 4G and 4H). EGFP-actin also distributed as patches on the surface at 4C (Figure 4G, arrows). At 37C, these patches migrated to form a spherical extrusion (Figure 4H). We hereafter refer to this spherical extrusion as “actin-rich extrusion.” The total amount of F-actin/cell, determined spectrofluorometrically using fluorescent phalloidin, did not change before and after treatment with bPEG-Chol/SA (data not shown). Furthermore, the actin-rich extrusion was completely abolished by latrunculin A or jasplakinolide, suggesting that polymerization/depolymerization of actin was required for the extrusion.
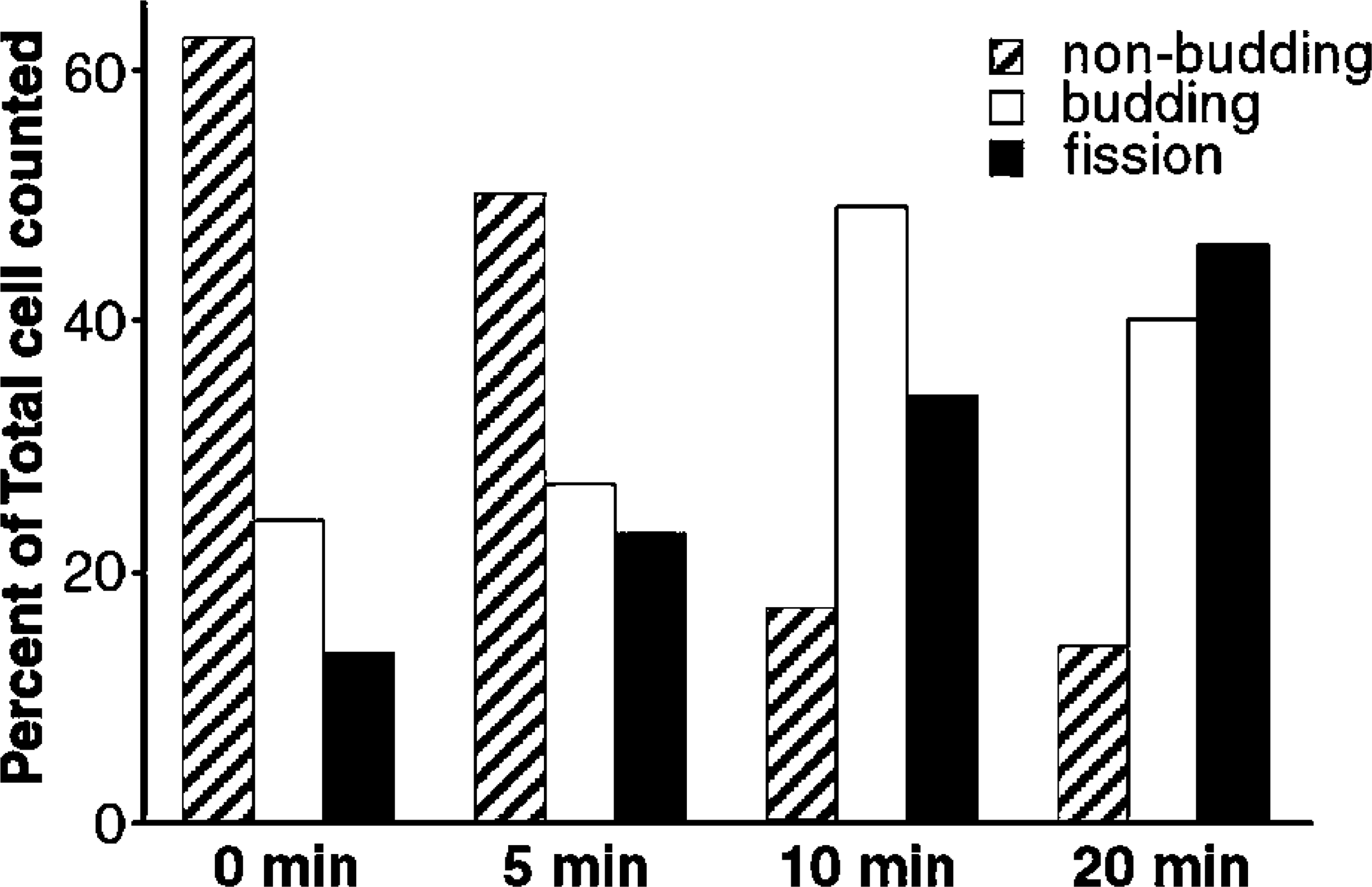
Histogram of cells exhibiting budding or fission of actin-rich extrusions. OKA-treated K562 cells were treated as in Figure 2. Cells were scored for non-budding (see Figure 2B), promoting budding (see Figures 2D and 2F), or fission (see Figures 2G and 2H).
Although the effect of SA was marked, we noted that bPEG-Chol alone induced an actin-rich extrusion in a few OKA-treated cells similar to those in Figures 2E and 4D. This change appeared to be induced by the shear stress that was applied during pipetting or mounting on a glass slide. Indeed, gently pressing the OKA/bPEG-Chol/SA-treated cells via coverslips induced the actin-rich extrusion even at 4C (data not shown). These results suggest that membrane stresses that deform cortical actin are similarly effective for bPEG-Chol/SA.
When OKA/bPEG-Chol/SA-treated cells were observed in an electron microscope, we observed a thick actin bundle (Figure 8B, small arrows, and Figure 9A). Near this structure, intermediate filaments also bundled (Figure 8B, asterisk). Moreover, mitochondria and some electron-lucent vesicles also accumulated near the actin-rich extrusion (Figures 8C and 8D). Surprisingly, the electron micrographs at higher magnification revealed accumulation of many clathrin-coated pits over the surface of the actin-rich extrusion (Figure 9B). They were homogeneously sized and open. By immunofluorescence, an AP-2 component, α-adaptin, had also accumulated (Figure 5A). In marked contrast, dynamin and the transferrin receptor (TfR) homogeneously distributed over the whole surface (Figure 5D and 5G). When TfR molecules were crosslinked by a combination of primary/secondary antibodies, only small patches were induced (data not shown). Moreover, distribution of an early endosomal protein, EEA1, also did not change (Figure 5J). These results suggested that TfR in OKA-treated cells are not linked to the cytoskeleton or to the early endosomal membrane.
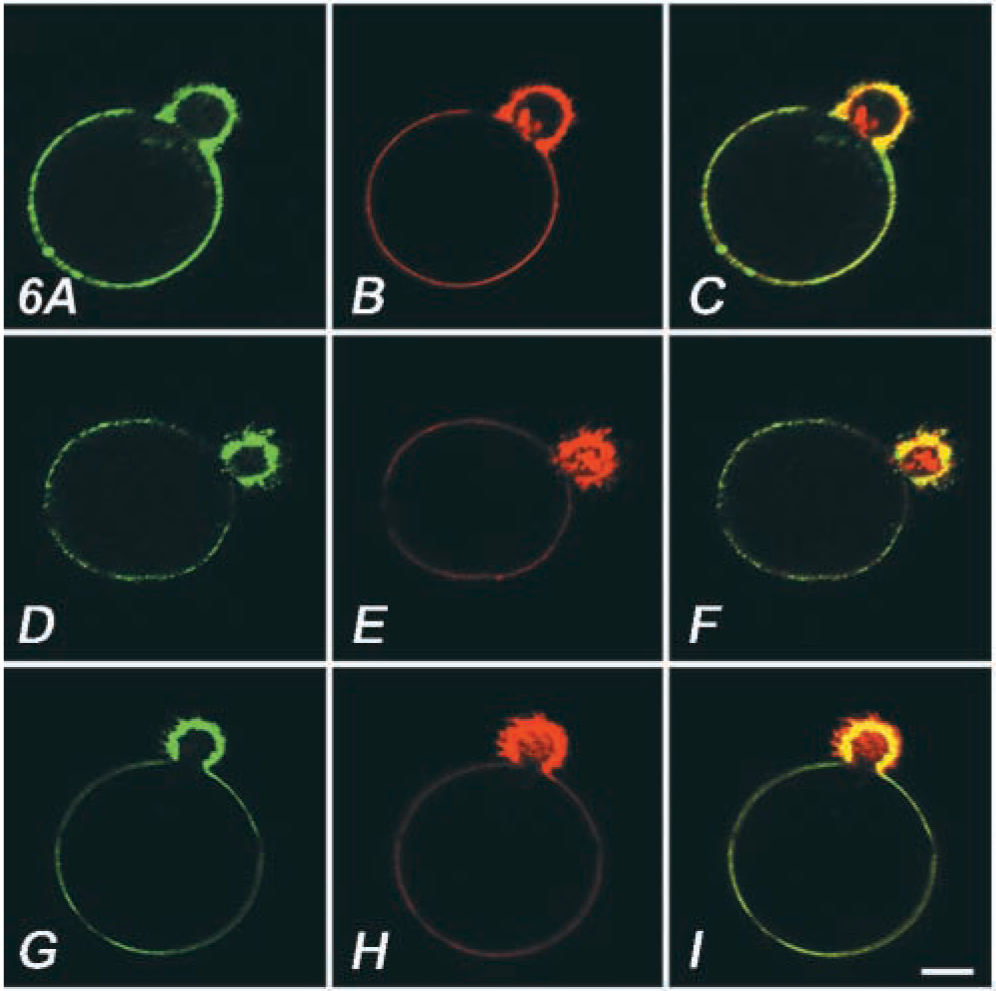
We examined the distribution of markers for lipid rafts, CD59, a typical GPI-anchored protein, and ganglioside GM1 (Figure 6). The molecules, visualized by an MAb and by cholera toxin B, respectively, distributed evenly over the cell surface at 4C (data not shown). In contrast, when the cells were incubated at 37C, both molecules were accumulated on the actin-rich extrusion (Figures 6D and 6G). Another protein, CD45, a putative protein tyrosine phosphatase segregated from an immunological synapse (Espinosa et al. 2002), showed very slight accumulation (Figure 6A). We next assessed the effect of modification of raft components. By crosslinking CD59, an actin-rich extrusion was induced in OKA-treated cells without bPEG-Chol/SA (Figures 7A–7C). Moreover, binding of integrin β1 by an MAb TS2/16, which potentiates the molecule for binding to cortical actin and ECM (van de Wiel-van Kemenade et al. 1992), also induced an actin-rich extrusion after crosslinking with a secondary antibody (Figures 7D–7F). In contrast, a non-activating MAb, DF5, did not induce an effect (Figures 7G–7I).
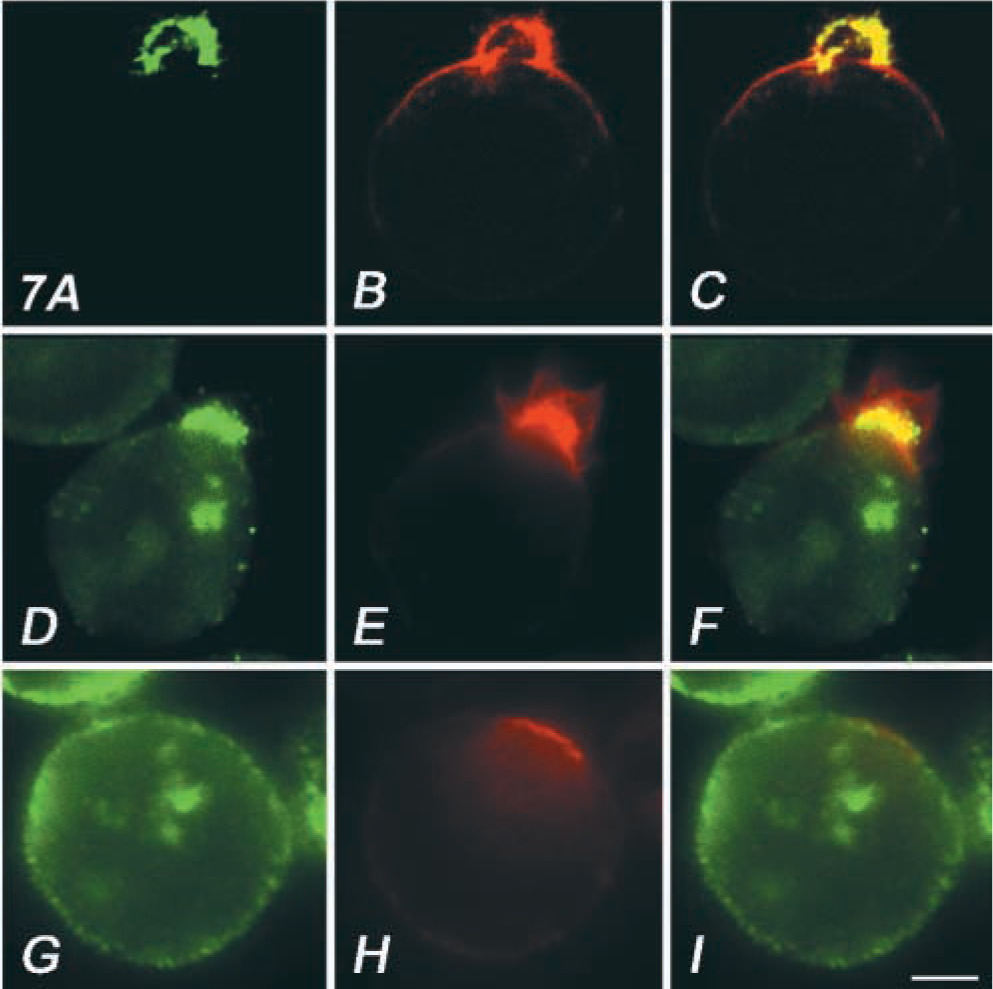
A non-receptor tyrosine kinase, fyn, regulates raft dynamics in T-cells (Bertagnolli et al. 1999; Harder and Simons 1999; Krause et al. 2000). In fyn(−/−) cells, we investigated the involvement of its activity in formation of the actin-rich extrusion. T-cell lines from normal and fyn(−/−) mutant mice were treated with OKA and then with bPEG-Chol/SA. As shown in Figures 10A and 10B, treatment of control fyn(+/+) cells induced spherical extrusion that was associated with thy-1, a lipid raft maker in T-cells. In contrast, the spherical extrusion formation was inhibited in fyn(−/−) cells (Figures 10C and 10D). These results indicate that fyn is one of the key tyrosine kinases in transducing the effect of OKA/bPEG-Chol/SA to the actin cytoskeleton.
Discussion
Treatment of cells with okadaic acid (OKA) is often described to mimic the cytoplasmic condition of mitotic cells. In particular, arrest of membrane traffic and cell motility in the cells has given the impression that cell activities are suppressed. However, we found that marked accumulation of actin was induced when cells were treated with bPEG-Chol and SA at 37C (Figures 4 and 8). Actin was similarly accumulated when a raft component, CD59, was crosslinked. Moreover, an experiment using MAb TS2/16, which activates the linkage between integrin β1 and actin, induced a similar response (Figure 7). Actin was also accumulated when the cells were directly compressed via a coverslip. In contrast, this response was not induced by the non-activating binding of anti-integrin MAb DF5 (Figures 7G–7I) or by anti-TfR MAb (data not shown). In a previous study on normal K562 cells, we found that even fourfold more PEG-Chol (20 nmol per 5 × 106 cells) did not induce accumulation of actin (Baba et al. 2001). These results indicate that OKA treatment induces a specific cellular condition that mobilizes whole cell actin to the cortex by biological as well as physical stimuli and establishes a tight coupling between actin and the membrane components.
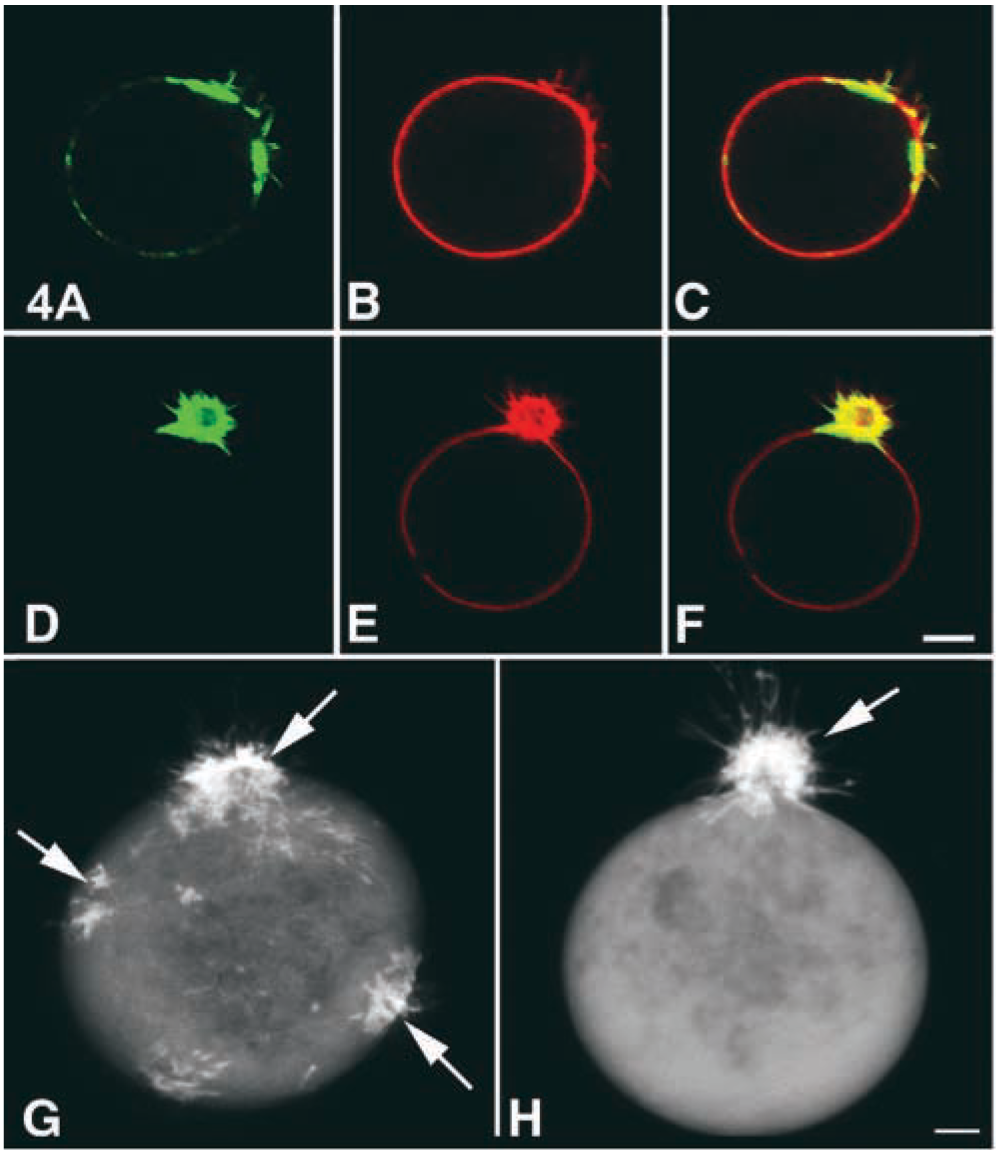
Confocal images of the distribution of F-actin. The cells were doubly stained for F-actin (
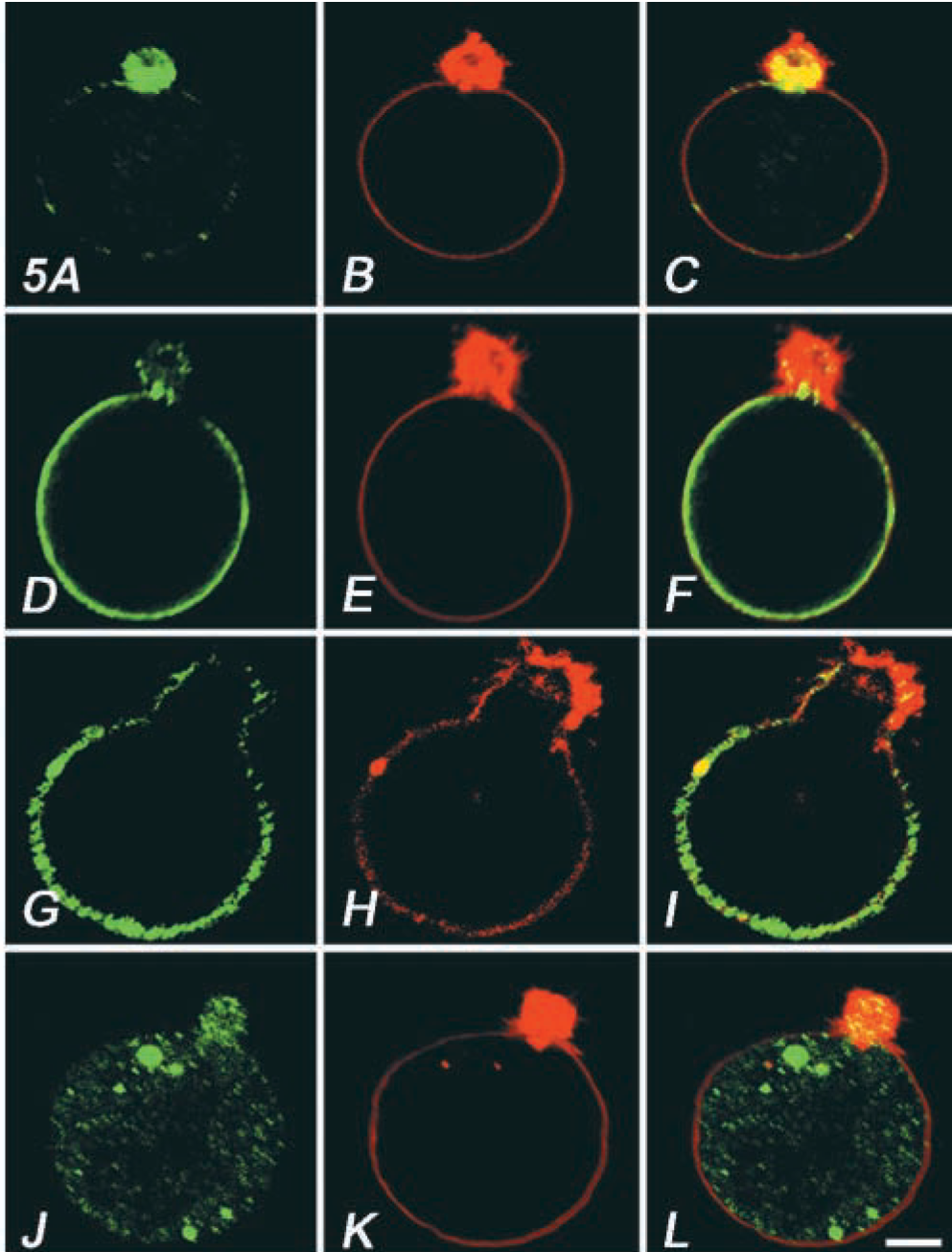
Distribution of endocytic protein markers, α-adaptin (
Distribution of CD45 (
Ligation of surface molecules with a specific antibody induced actin-rich extrusion. OKA-treated K562 cells were incubated with anti-CD59 MAb (
By summarizing the microscopic results on bPEG-Chol/SA-treated cells along a time sequence, the following scenario can be drawn. (a) OKA induces actin to underline certain plasma membrane domains (Figure 4G). (b) When the surface is stimulated, these actin-bound patches move into a single pole (Figures 4D–4F and 4H). (c) Mobilization to the cortex is propagated to the augmentation of perinuclear intermediate filament-containing actin (Figures 8A and 8B). (d) Lipid raft or clathrin-coated pit components are mobilized with the accumulated actin during steps b and c. Mitochondria and some other membrane compartments, but not EEA1-associated early endosomes, are also mobilized (Figures 5, 8, and 9). (e) Concentration of these elements in a single extrusion occurs, followed by separation from the enucleated part (Figures 2G and 2H). We believe that each of these reaction steps is important for evaluating the molecular linkage between actin and specific sets of membrane domain elements. Analysis of T-cell lines using gene-knockout mice indicated involvement of non-receptor kinase fyn in OKA/bPEG/SA-induced cell extrusion (Figure 10). In the cells, not only actin but also a raft marker, Thy-1, failed to accumulate. In the presence of active fyn, aggregation of rafts induces association of actin filaments together with many tyrosine-phosphorylated proteins (Harder and Simons 1999). In T-cells, crosslinking of CD44 induces recruitment of CD44 and of lck and fyn into the raft-like membrane fractions (Foger et al. 2001). Taken together, these points suggest that one candidate mechanism may be that clustering of PEG-Chol-embedded domains triggers recruitment of fyn to rafts. Studies assessing the distribution of bPEG-Chol in the rafts are now being undertaken.
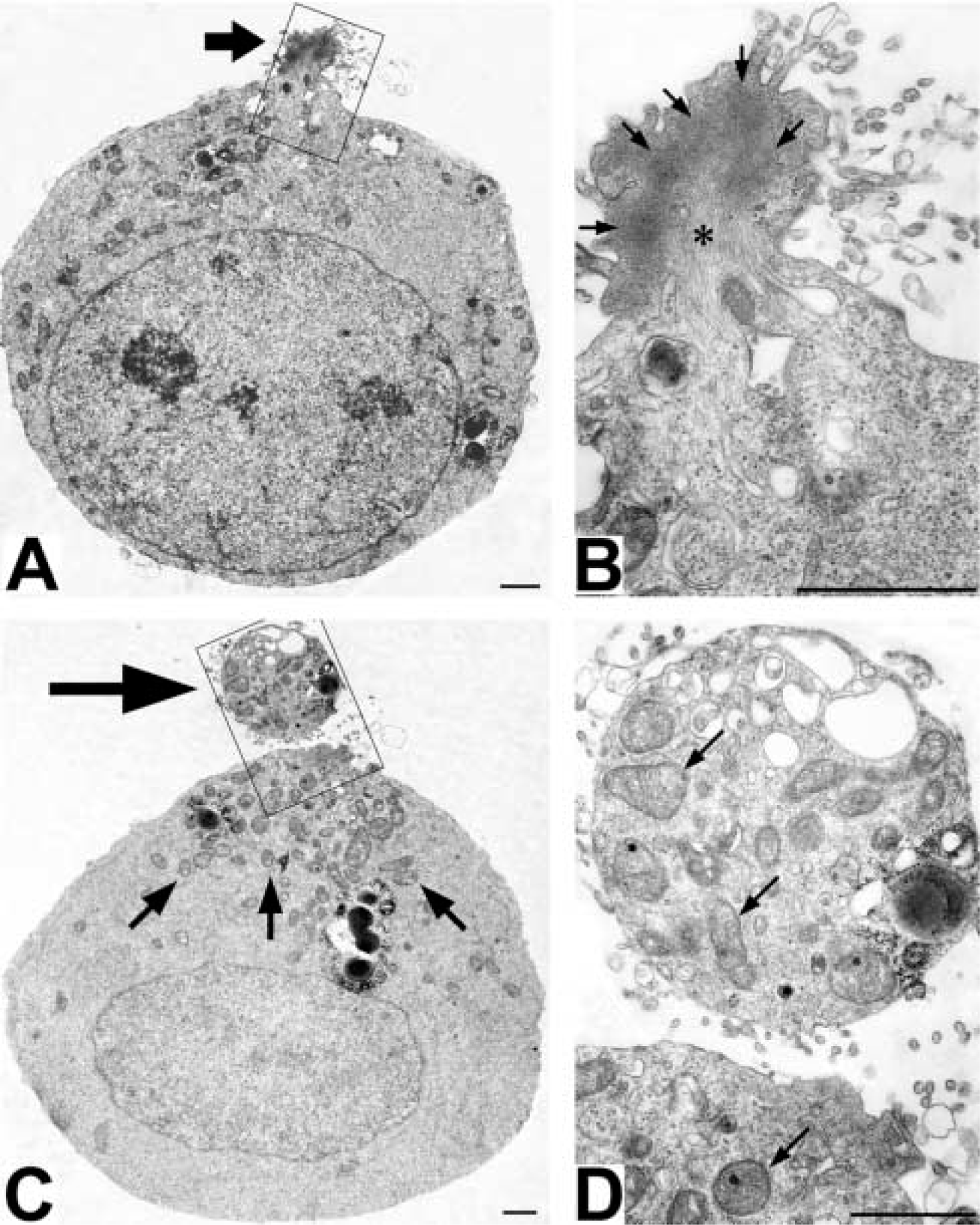
Electron micrographs of OKA/bPEG-Chol/SA-treated K562 cells. K562 cells treated with 500 nM OKA at 37C for 30 min were further treated with bPEG-Chol/SA and incubated at 37C for 20 min. In an ultrathin section, accumulated F-actin protruded at the extruding body (
Formation of the actin-rich extrusion involved mobilization of the perinuclear cytoskeleton. Our observations indicated that vimentin, which initially scattered as clusters around the nucleus, assembled in a bundle due to bPEG-Chol/SA (Figures 8B and 9B). Disorganization of the perinuclear cytoskeleton by phosphorylation of vimentin has been suggested for adherent cells by a number of studies. In particular, inhibitors of protein dephosphatases, OKA (Lee et al. 1992), mycrocystin-LR (Khan et al. 1996), and calyculin A (Hirano et al. 1992) are very potent inducers. One of these authors reported that the treatment induced collapse of the perinuclear actin network and subsequent formation of an actin “ball” in NIH3T3 with calyculin A (Hirano et al. 1992). In the cells, the ball and the nucleus were connected via vimentin-containing intermediate filaments. Our preliminary experiments indicated that K562 cells treated with 100 nM calyculin A and bPEG-Chol/SA similarly induced separation of the actin-rich extrusion (unpublished results). The perinuclear organization of OKA-treated K562 cells therefore appears to be very similar to these cells.
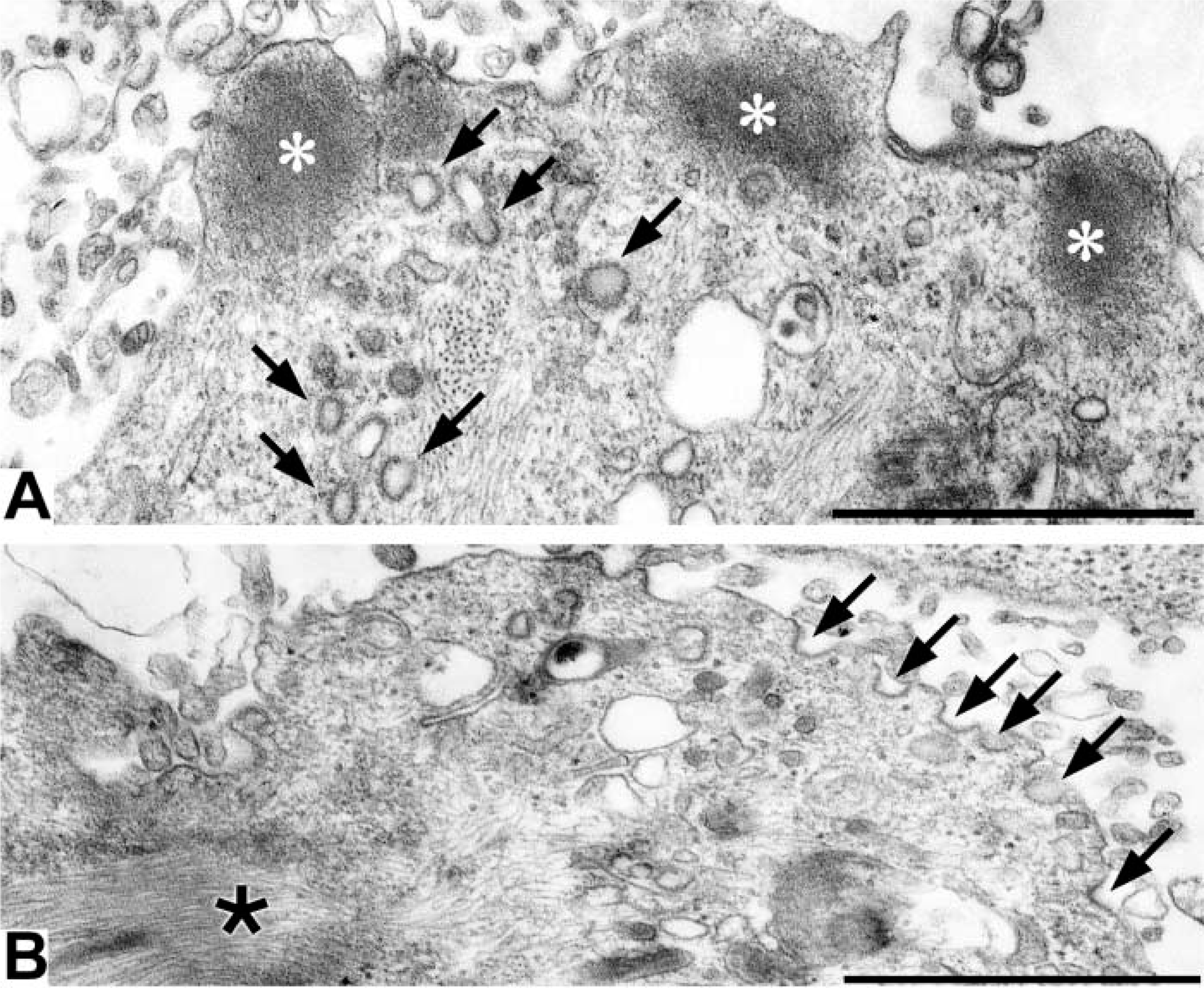
Electron micrographs of spherical extrusion. Various endocytic structures, such as endosomes (
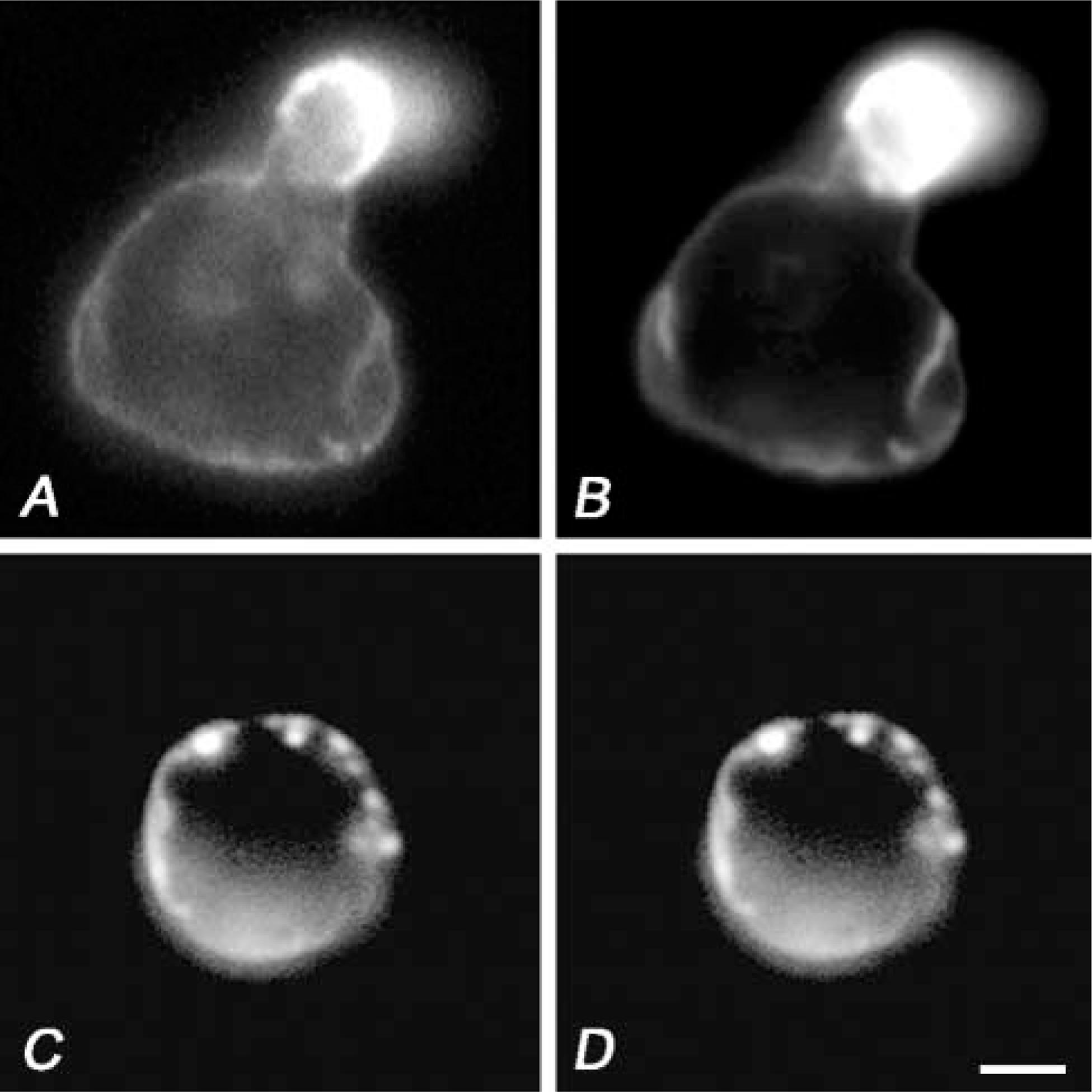
A spherical extrusion was not induced in an fyn(−/−) murine T-cell. Immunolocalization of Thy-1 (
In this study we used PEG-Chol to address the coupling of cortical actin with membrane microdomains. In comparison to the commonly used reagent methyl β-cyclodextrin, which had effects not only on lipid rafts but on clathrin-coated pits and integrity of the plasma membrane (Subtil et al. 1999), our approach using PEG-Chol-SA crosslinking had the advantage that the degree of membrane stress can be quantitatively controlled by changing the applied amount of PEG-Chol. Our approach, combined with cytoskeletal modifications, is potentially useful for elucidating the links between membrane microdomains and cytoplasmic organization.
Footnotes
Acknowledgements
SBS and TB contributed equally to the research. Supported by grants to SBS and TB from the Japanese Ministry of Education, Culture, Sports and Science.
We thank Drs Koichiro Miyajima (Osaka University of Pharmaceutical Sciences) and Yoshio Hamashima (Kyoto Pharmaceutical University) for PEG-Chol and bPEG-Chol, and Dr Yoshinori Fujiyoshi (Kyoto University) for support and discussion.