
Abstract
A general problem in immunocytochemistry is the development of a reliable multiple immunolabeling method when primary antibodies must be used that originate in the same species. We have developed a protocol for the immunodetection of three antigens in a single tissue preparation, using unconjugated primary antibodies raised in the same species. Immunocytochemical detection of neuronal nitric oxide synthase, calcitonin gene-related peptide, and calbindin D28k in the lung of rats demonstrated that part of the pulmonary neuroepithelial bodies are selectively contacted by at least three different nerve fiber populations. The first antigen was detected using tyramide signal amplification, a very sensitive method allowing a dilution of the first primary antibody far beyond the detection limit of fluorescently labeled secondary antibodies. The second antigen was visualized by a fluorophore-conjugated secondary monovalent Fab antibody that at the same time blocks the access of the third secondary antibody to the second primary antibody. Moreover, the monovalence of the Fab fragment prevents the third primary antibody from binding with the second-step secondary antibody. The triple staining technique described here is generally applicable, uses commercially available products only, and allows the detection of three antigens in the same preparation with primary antibodies that are raised in the same species.
Keywords
T
A general problem in ICC relates to the fact that most of the available primary antibodies originate mainly from two species, i.e., rabbit and mouse. Consequently, several methods to achieve double immunostaining with primary antibodies raised in the same species have been developed. The first true double labeling protocols required antibody elution techniques (for review see Larsson 1988), which are not always reliable and occasionally damage tissues, especially those containing a high amount of loose connective tissue, as is the case for lungs. An alternative approach was described by Wang and Larsson (1985), who found that formaldehyde vapor treatment inactivates the free anti-IgG binding sites of fluorophore-labeled secondary antibodies, thereby saturating antigenic epitopes of first layer primary antisera. However, this method is associated with the risk of rendering formaldehyde-sensitive antigens undetectable in the following steps.
Recently, a few more consistent methods have been described for using antibodies raised in the same species in double immunofluorescence cytochemistry. Tornehave et al. (2000) showed that moderate micro-waving between the first and second staining cycles prevents the reaction of the antibodies present in the first step with subsequently applied reagents. Kroeber et al. (1998) developed a technique by which free binding sites of the first secondary antibody were blocked by normal serum and by which the second secondary antibody was coupled to the second primary antibody in a reaction tube. Shindler and Roth (1996) developed a two-step technique employing detection systems of different sensitivities. In the first step they used the very sensitive tyramide signal amplification (TSA) to detect a highly diluted first primary antibody. In the subsequent conventional immunostaining method, the secondary antibody failed to detect the primary antibody from the first step.
An elegant double staining method is the use of fluorochrome-conjugated monovalent Fab fragments as secondary antibodies, as described by Negoescu et al. (1994). In this approach the secondary polyclonal monovalent Fab fragments used to visualize the first primary antibody prevent the secondary antibodies, used to detect the second primary antibodies, from binding to the first primary antibody. The monovalence of the Fab fragments also eliminates the possibility of linking the primary antibody from the second step. Alternative approaches utilized unlabeled monovalent Fab fragments to block residual binding sites on tissue-bound antibodies processed in conventional procedures (Lewis Carl et al. 1993) or to build multilayer detection systems (Donat et al. 1999).
In the present investigation, simultaneous labeling of multiple antigens was applied to study the spatial relationship between different nerve fiber populations contacting pulmonary neuroepithelial bodies (NEBs) in rats. Pulmonary NEBs are clusters of neuroendocrine cells present in the epithelium of all air-breathing vertebrates (for reviews see Sorokin and Hoyt 1989; Adriaensen and Scheuermann 1993; Sorokin et al. 1997; Cutz and Jackson 1999). In view of their characteristic location, receptor-effector-like morphology, and complex innervation, NEBs may play a major role in the local regulation of airway function. Our previous investigations have provided evidence for at least two different extrinsic sensory nerve fiber populations (Adriaensen et al. 1998; Brouns et al. 2000b) and one intrinsic nerve fiber population (Brouns et al. 2000b; Adriaensen et al. 2001) that selectively contact pulmonary NEBs in rats. Nerve tracing experiments (Adriaensen et al. 1998), denervation studies, and ICC staining confirmed that NEBs in rat lungs receive an extensive intraepithelial vagal nodose calbindin D28k (CB)-immunoreactive (IR) but calcitonin gene-related peptide (CGRP)-negative sensory nerve fiber population, in addition to the CGRP-IR afferent nerve fiber population described for many years in connection with pulmonary NEB cells (Cadieux et al. 1986; Shimosegawa and Said 1991; Sorokin et al. 1997) and which appears to form basal contacts only (our unpublished observations). Using double immunolabeling, we have recently demonstrated the presence of a third intrinsic nerve fiber population expressing neuronal nitric oxide synthase (nNOS), which also appears to form extensive nerve terminals between the neuroendocrine cells of pulmonary NEBs (Brouns et al. 2000b; Adriaensen et al. 2001).
Multiple immunofluorescence labeling in combination with confocal laser scanning microscopy is a powerful strategy to visualize the spatial relationship between different antigens in histological sections. However, previous experiments have shown that an appropriate combination of antibodies raised in different species against nNOS, CGRP, and CB as markers for distinct nerve fibers that selectively contact NEBs in rat lung is not available. Therefore, a triple immunofluorescence labeling method was developed using three commercially available polyclonal antibodies raised in rabbit against nNOS, CGRP, and CB. The method described here is based on the use of TSA enhancement for detection of the first antibody, fluorophore-labeled and unlabeled Fab secondary antibodies for the second antibody, and standard fluorophore-labeled secondary antibodies for the third primary antibody.
Materials and Methods
Lungs of 10-day-old Wistar rats (n = 4; Iffa Credo, Brussels, Belgium) were used. Procedures were approved by the local ethics committee of the University of Antwerp and conformed to European Community regulations.
Animals were sacrificed by IP injection of an overdose of sodium pentobarbital (Nembutal). Lungs were intratracheally instilled with Zamboni's solution (0.2% saturated picric acid and 4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4), dissected, de-areated, and fixed in the same solution for 30 min. After a rinse in PBS, 0.01 M, pH 7.4, lungs were treated for improvement of the ICC conditions (Llewellyn-Smith et al. 1985), stored overnight (ON) in 20% sucrose (in PBS; 4C), and mounted in Tissue Tek (Sakura Finetek Europe; Zoeterwoude, The Netherlands). Thirty-μm-thick cryostat sections were thaw-mounted on poly-
All ICC incubations were carried out at room temperature in a humidified container. Unless indicated otherwise, washes with PBS were performed between each step, and all primary and secondary antibodies were diluted in PBS containing 10% normal goat serum, 0.1% bovine serum albumin, 0.05% thimerosal, and 0.01% NaN3 (PBS+).
For detection of the first primary antibody, a biotin-conjugated tyramide signal amplification (TSA) kit (NEL700; Perkin-Elmer Life Sciences, Boston, MA) was applied. Because tyramide deposition in the immediate neighborhood of the antigen is catalyzed by the presence of horseradish peroxidase (HRP), endogenous peroxidase activity of freeze-sections was first blocked by H2O2 (0.4% in 50% methanol in PBS, 10 min). To minimize nonspecific binding of all secondary antibodies raised in goats and to enhance penetration of antisera, preincubation was performed for 30 min with PBS+ containing 1% Triton X-100. In the first primary incubation, lung sections were incubated with a rabbit polyclonal antibody against rat nNOS (diluted 1:20,000; ON; Euro-Diagnostica, B220–1, Malmö, Sweden). Sections were consecutively incubated with biotin-conjugated Fab fragments of affinity-purified goat anti-rabbit immunoglobulin (Ig)G (H+L) (diluted 1:2000; 1 hr; Rockland, 811–1602, Gilbertsville, PA) and ExtrAvidin-HRP (diluted 1:2000 in PBS containing 0.05% thimerosal and 0.1% BSA, 1 hr; Sigma E2886, St Louis, MO). Between subsequent steps, sections were washed in PBS containing 0.05% Tween-20. Sections were then incubated for 10 min with biotin-conjugated tyramide diluted 1:100 in “amplification solution.” Visualization was performed using Cy3-conjugated streptavidin (diluted 1:6000 in PBS, 10 min; Jackson 016-160-084, West Grove, PA).
To allow combination with the second and third primary antisera, both raised in the same species, further immunostaining was performed according to the method described by Negoescu et al. (1994). The nNOS-labeled cryostat sections were incubated with a second rabbit polyclonal antibody raised against CGRP (diluted 1:200; ON; Affiniti CA1134, Exeter, UK), followed by a fluorescein (FITC)-conjugated Fab fragment of goat anti-rabbit IgG (H+L) (diluted 1:100, 6 hr; Jackson 111-097-003). After rinsing, sections were incubated with unlabeled Fab fragments of goat anti-rabbit IgG (H+L) (diluted 1:100, 3 hr; Jackson 111-007-003) to block all possible remaining binding sites of the second primary antibody.
In the third step, sections were incubated with a third rabbit polyclonal antiserum directed against CB (diluted 1:2000, ON; SWant CB-38, Bellinzona, Switzerland). This primary antibody was detected with a Cy5-conjugated goat anti-rabbit IgG (diluted 1:500, 1 hr; Jackson 111-175-144). After a final wash, sections were mounted in Vectashield (Vector Laboratories; Burlingame, CA).
Negative staining controls for the three ICC procedures were performed by substitution of non-immune serum for the primary or secondary antisera. To obtain a clear interpretation and to check for non-crossreactivity after consecutive staining with the three rabbit primary antisera, the results of single and double immunostaining were evaluated for all combinations of antigens and compared with those from triple labeling. For the TSA-enhanced procedure, staining controls were performed by omission of the primary antiserum of either the second or third incubation, or both. A non-amplified staining with antibodies against nNOS, using the same concentration (1:20,000) as for the TSA-enhanced reaction, was routinely included. To compare TSA-enhanced and conventional immunostaining, additional sections of rat lung were subjected to conventional immunostaining with antibodies against nNOS diluted 1:100.
As controls for crossreaction of the second and third immunolabeling, interference control stainings for the use of Fab fragments were performed as described by Negoescu et al. (1994). As an additional positive control, the second and third labeling were reversed in a double labeling procedure. Therefore, cryostat sections were consecutively incubated with rabbit anti-CB (diluted 1:2000, ON), FITC-conjugated Fab fragments of goat anti-rabbit IgG (H+L) (diluted 1:100, 6 hr), unlabeled Fab fragments of goat anti-rabbit IgG (H+L) (diluted 1:100, 3 hr), rabbit anti-CGRP (diluted 1:500, ON) and Cy5-conjugated goat anti-rabbit IgG (diluted 1:500, 1 hr).
To obtain detailed images of the individual nerve endings in contact with NEBs, a confocal laser scanning microscope (Zeiss LSM 510) equipped with image reconstruction facilities (Imaris 2.7 software; Bitplane, Zürich, Switzerland; Silicon Graphics Indigo 2 workstation) was used. An argon laser (488 nm) and two helium-neon lasers (543 nm and 633 nm) were utilized for the excitation of FITC, Cy3, and Cy5, respectively.
Results
Triple immunostaining with rabbit antibodies against nNOS, CGRP and CB clearly demonstrated that some pulmonary NEBs in rat lungs are selectively contacted by at least three different nerve fiber populations. Most eye-catching were nNOS-IR nerve fibers that ramify at the base of the epithelium and give rise to terminal arborizations between the epithelial cells (Figures 1A and 2). Combined detection of nNOS and CGRP, CB, or both revealed the absence of CGRP and CB immunoreactivity from nNOS-IR nerves. Pulmonary NEB cells express both CB and CGRP, although the intracellular staining pattern of both substances differs (Figures 1B, 1C, and 2). In the neighborhood of NEBs, CGRP-IR varicose nerves are seen to run in close proximity to the nitrergic fibers, often requiring confocal microscopy for discrimination between both nerve fiber types (Figures 1 and 2). Thick CB-IR nerve fibers contacting pulmonary NEBs are not IR for CGRP or nNOS (Figures 1 and 2).
Comparison of the triple staining protocol (Figures 1 and 2) and the reversed double control staining for CB and CGRP did not yield any differences in the localization of CGRP and CB IR. Single labeling for the three rabbit antibodies in different sections showed results very similar to the separate images for the three antibodies in triple labeled sections. Immunofluorescence was not observed in the negative controls of single incubations. No reactions were observed for the second and third labeling after omission of the appropriate primary antibodies. After consecutive double or triple labeling, nNOS IR appeared identical to that seen in single TSA-enhanced nNOS immunostaining. Non-amplified conventional indirect nNOS immunostaining using a 1:100 dilution of the primary antibody did not reveal nNOS-IR nerve terminals that contact NEBs in rat lungs, while nNOS-expressing neuronal cell bodies were weakly labeled. Non-amplified indirect immunolabeling using the same concentration (1:20,000) of primary antiserum against nNOS as in TSA-enhanced reactions did not result in detection of nNOS-IR nerve terminals. Interference control stainings showed that there was neither linking of the third-step secondary antibody with the second-step primary antibody nor of the third-step primary antibody with the second-step secondary antibody.
Discussion
The present work describes a reliable triple immuno-fluorescence labeling procedure using unconjugated primary polyclonal antibodies raised in the same species. All products used are commercially available. The simultaneous detection in a single preparation of nNOS, CGRP, and CB in three different nerve fiber populations that selectively contact pulmonary NEBs is illustrated and accords with our recent observations obtained by double labeling procedures (Brouns et al. 2000b; Adriaensen et al. 2001). nNOS IR was visualized using TSA enhancement (Shindler and Roth 1996), followed by detection of CGRP by a fluorophore-labeled Fab secondary antibody (Negoescu et al. 1994) and subsequent detection of CB with a fluorophore-labeled “conventional” secondary antibody. All possible remaining binding sites of the second primary antibody were blocked using unlabeled anti-rabbit Fab fragments (Lewis Carl et al. 1993) between the second and third steps.
TSA or catalyzed reporter deposition (CARD) amplification, introduced by Bobrow et al. (1989), is based on the deposition of complexes of labeled tyramide around peroxidase molecules without interfering with subsequent ICC procedures. The optimal dilution of primary antiserum for detection by TSA has been reported to be up to 200 times higher than in standard staining protocols (Adams 1992). Such high dilutions render most primary antisera virtually undetectable by conventional fluorophore-conjugated secondary antibodies. On this basis, TSA allows the consecutive application of two antisera raised in the same species, provided that the concentration of the first (enhanced) primary antibody in the preparation is too low to be detected by subsequent non-amplified procedures (Shindler and Roth 1996). Moreover, previous investigations (Shindler and Roth 1996; Brouns et al. 2000a) clearly demonstrated that the detectability of co-localized antigens is not impaired by prior deposition of tyramide.
In the present study, TSA-enhanced ICC labeling for nNOS and subsequent application of FITC-labeled Fab fragments of anti-rabbit secondary antibodies, or Cy5-labeled anti-rabbit antiserum, did not result in detectable binding of these secondary antibodies to the nNOS-IR nerve fibers. Moreover, nNOS IR did not co-localize with CB or CGRP IR, clearly illustrating that the fluorophore-conjugated secondary antibodies applied in the second and third step of the protocol were not able to detect the highly diluted, initially bound rabbit anti-nNOS antibody.
The most significant advantage of TSA enhancement is that several antibodies that are considered non-reactive in conventional staining protocols show strong and reproducible immunoreactivity using TSA amplification. The necessity to use TSA enhancement for a clear visualization of intraepithelial nNOS-IR nerve terminals was set by the fact that non-amplified immunostaining (dilution of primary antibody 1:100) failed to label intraepithelial nitrergic nerve terminals in 10-day-old rats, whereas intrapulmonary nitrergic neuronal cell bodies were weakly stained. Therefore, detection of the nNOS-IR terminals that contact NEBs in our application requires TSA enhancement and is determining for the sequence of the primary antibodies in the triple immunofluorescence staining protocol. When primary antibodies are used that all work well in single conventional immunostainings, one of them can be randomly chosen to be used in the first TSA-enhanced step at significantly higher dilutions (King et al. 1997).
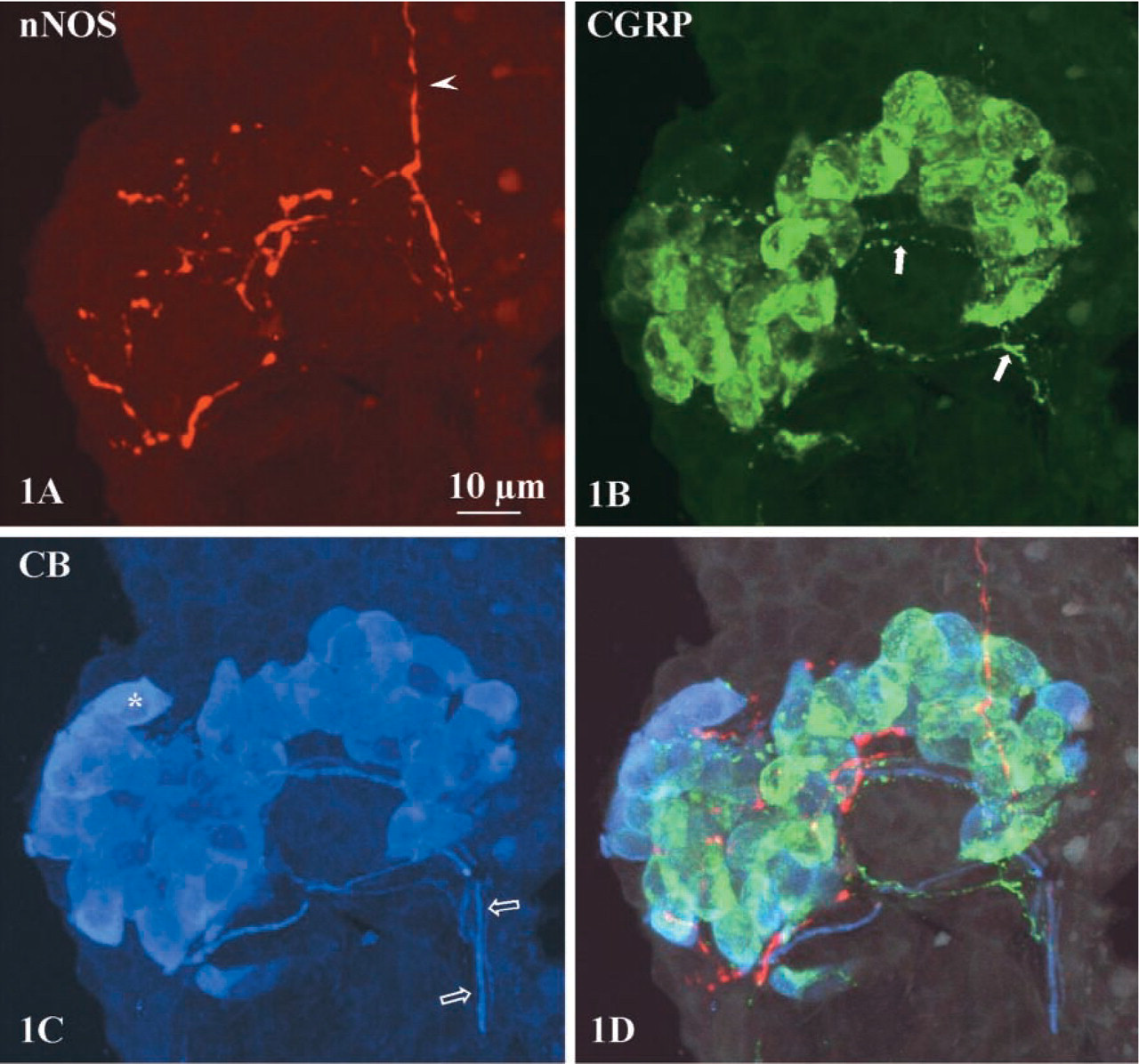
Maximal intensity projections of 30 confocal optical sections (1-μm interval) of an NEB in a bronchiole.
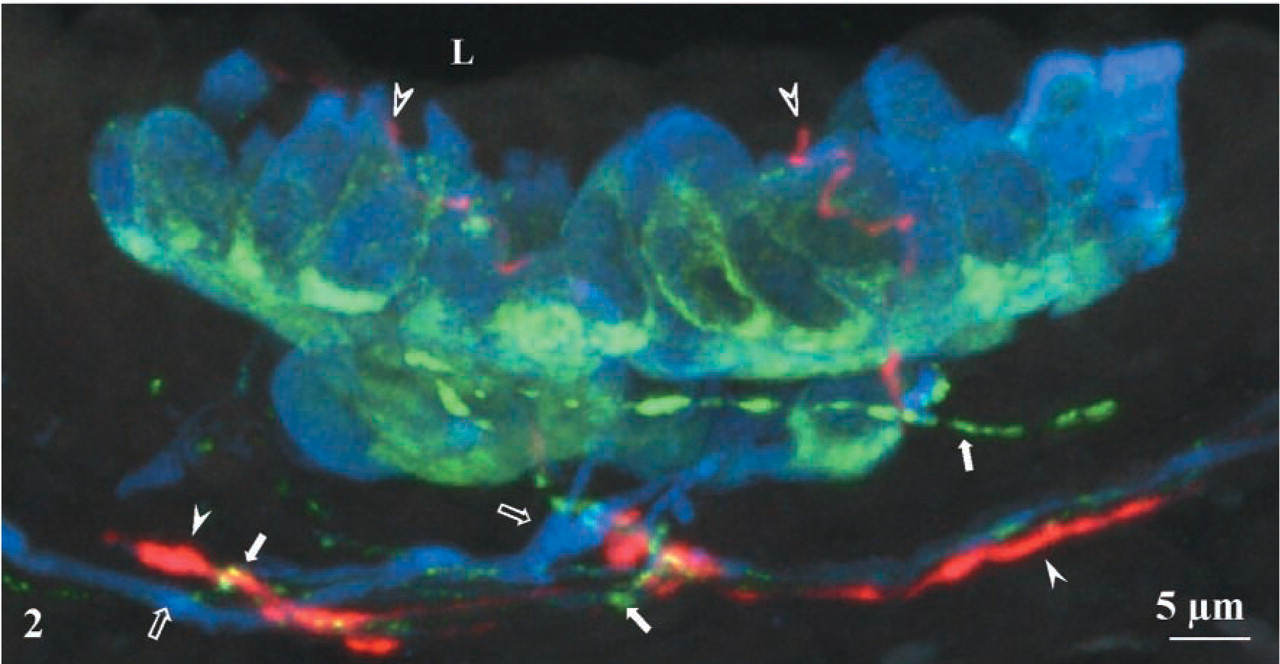
Maximal intensity projection of 33 confocal optical sections (1-μm interval). A bronchial group of CGRP/CB-IR NEB cells is contacted by three different nerve fiber populations. Thick blue CB-IR nerves (open arrows) present in the lamina propria of the airway, branch, and innervate the NEB. Thin varicose green CGRP-IR nerve fibers (arrows) contact the NEB at its basal side. Red nNOS-IR nerve fibers (arrowheads) run in direct proximity to CGRP-IR fibers in the lamina propria, but penetrate between the CGRP/CB-IR neuroendocrine cells, apparently almost reaching (open arrowheads) the airway lumen (L).
The validity of the visualization of the third rabbit antibody in the proposed procedure is based on the use of a polyclonal monovalent Fab antibody for detection of the second rabbit antibody, thus preventing access of the third secondary antibody to the second primary antibody. Moreover, the monovalent structure of the Fab fragment renders capture of the third primary antibody impossible. Fine varicose CGRP-positive but CB-negative nerve fibers, as well as thick CB-positive but CGRP-negative nerve fibers, were seen to contact NEB cells in this study. The staining pattern of CB- and CGRP-IR nerve fibers and of CGRP/CB-IR NEB cells was identical in the triple staining procedure, in which CB was used as the third primary antibody and was detected using Cy5-conjugated secondary antibodies, and in the reversed control staining, in which CB was applied as the second rabbit antibody and detected using FITC-conjugated Fab fragments of anti-rabbit antibodies. The use of an additional step with unlabeled Fab fragments of anti-rabbit antibodies is based on a method by which fluorophore-labeled whole IgG secondary antibodies applied in a first step are blocked using normal serum and unlabeled monovalent Fab fragments (Lewis Carl et al. 1993). In the present triple staining protocol, the method of blocking access of the third-step secondary antibodies to the second-step primary antibodies (Negoescu et al. 1994) is combined with the method of using unlabeled Fab fragments (Lewis Carl et al. 1993), thereby providing a double security against binding of the third secondary antibody to primary antibodies applied in earlier steps.
The combination of three rabbit polyclonal antibodies, directed against nNOS, CGRP and CB, in this study can be used as the basis for a general triple-staining protocol for the application and detection in the same preparation of three polyclonal or monoclonal antibodies raised in the same species (see schematic in Figure 3). Because the proposed triple immunofluoresence staining method fully depends on blocking the access of the detection methods applied in the second and third steps of the staining protocol to the reagents used in the first and second steps, respectively, a number of prerequisites should be fulfilled before employing the method. (a) The use of TSA enhancement for visualization of the first primary antibody requires titration of the primary antiserum dilution beyond the detection limit of the second and third secondary antibodies (Shindler and Roth 1996) in such a way that binding of the first antibody to its antigen cannot be detected without an amplification step (Hunyady et al. 1996). (b) To achieve complete blockage and coverage of the epitopes on the primary antibodies, the Fab fragments used in the second step must originate from a polyclonal antibody (Negoescu et al. 1994). (c) Comparable with double labeling studies (Negoescu et al. 1994), the working conditions, i.e., Fab and other antibody concentrations and incubation times, must be carefully adapted to the specific antibody assembly of interest. Finally, excitation and emission spectra of the three fluorophores should permit selective visualization of the results.
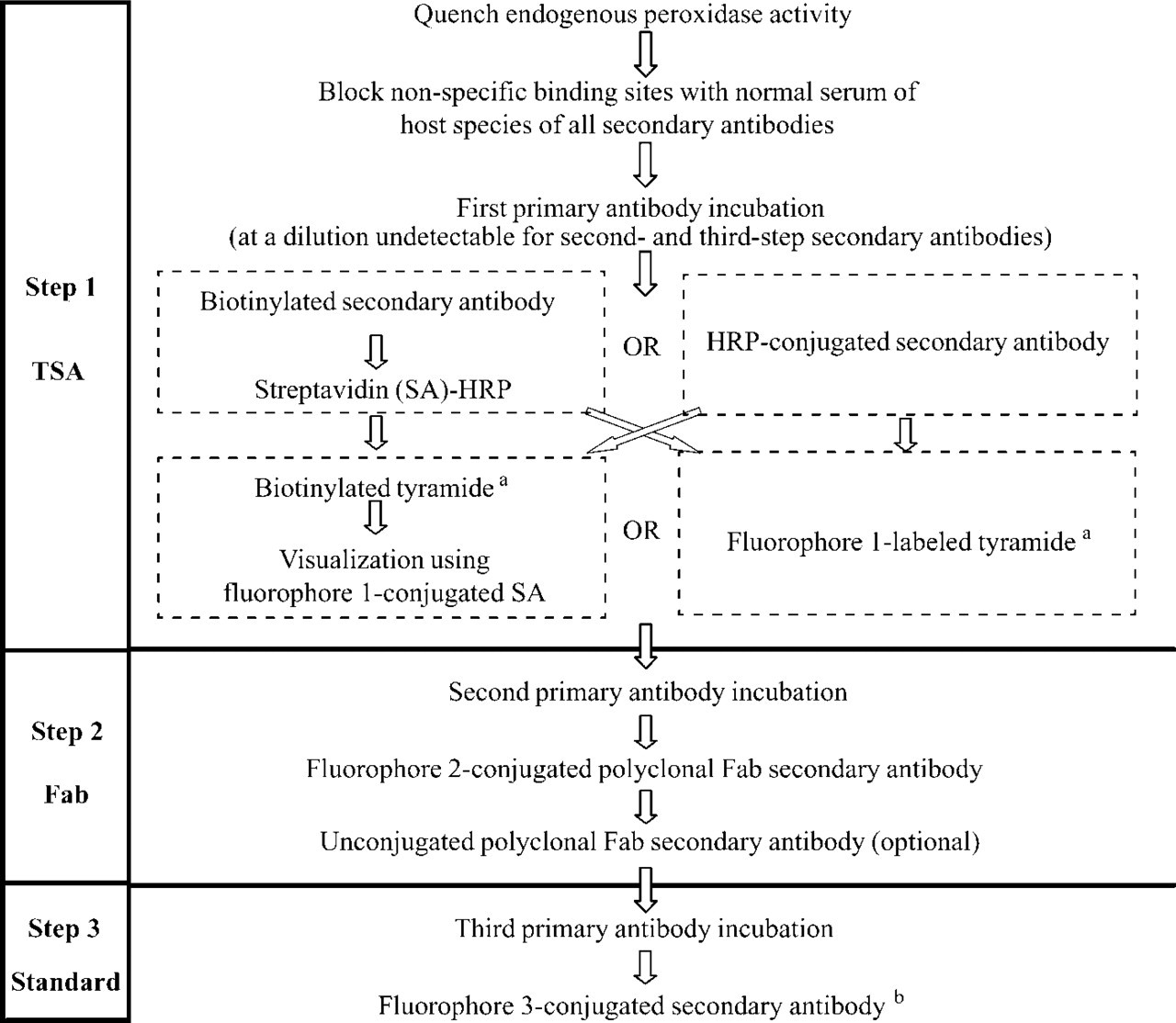
General protocol for immunofluorescence triple staining with antibodies raised in the same species. Incubation times and antibody dilutions are dependent on the antibody assembly. aCommercially available from Perkin-Elmer Lifesciences as TSA or TSA Plus; bIf an HRP-conjugated secondary antibody and the fluorophore 1-labeled tyramide are used for TSA enhancement in the first step, the fluorophore 3-conjugated secondary antibody may be replaced by a biotinylated secondary antibody and a consecutive fluorophore 3-conjugated SA.
In conclusion, the triple immunofluorescence staining procedure presented in this study does not require chemical alteration, masking, or eluting of primary antibodies, procedures that require additional steps which are time-consuming and may reduce detection sensitivity and selectivity. Because the primary antibodies are used in an unconjugated form and are detected by commercially available detection systems only, this staining protocol can easily be adapted to any trio of primary antibodies originating from the same species and is accessible to all investigators.
Footnotes
Acknowledgements
Supported by an IWT grant (SB 981363 to IB) and an FWO grant (G.0155.01 to DWS) from the Flemish Government, and by an NOI-BOF grant from the University of Antwerp.
We wish to thank L. Svensson and D. Vindevogel for technical assistance.