
Abstract
The aim of this study was to develop a model for the detection of individual cell adhesion molecules (CAMs) in the glycocalyx of spread human platelets using high-resolution cryo-field emission scanning electron microscopy (cryoFESEM). Three surface glycoprotein CAMs, P-selectin (CD62P), GPIba in the GPI-IX complex (CD42a/CD42bα,bβ), and the integrin GPIIbIIIa (CD41/CD61) in the human platelet were selected on the basis of their unique topographic shape. Spread human platelets were indirectly immunolabeled with 10-nm colloidal gold and then cryoimmobilized. After sublimation of water from the cryoimmobilized sample, partially freeze-dried platelets were coated unidirectionally with Pt, stabilized with carbon, and examined in an in-lens cryoFESEM using high-resolution back-scattered electron imaging. CAMs were detected by indirect immunogold labeling and the length of each type of CAM was determined using analysis of differences in parallax as measured in the software program Sterecon. Our results demonstrate the efficacy of using high-resolution cryoFESEM to recognize and detect individual CAMs in the glycocalyx. Further advances in production of metal coatings with finer granularity, together with improvements in imaging (tilting and angle of stereo images), may provide better definition of the topography associated with glycosylation and formation of multimeric CAM complexes.
Keywords
C
Ultrastructural spatial distribution of cell surface antigens on leukocytes has been accomplished by immunogold localization using field-emission scanning electron microscopy (FESEM) (Erlandsen et al. 1993; Hasslen et al. 1996). In these and other studies using immunogold labeling to study surface CAMs, the spatial distribution of the CAM has been inferred from the presence of the marker complex (immunogold), but little progress has been made in detection and recognition of individual CAMs directly. Development of cryotechniques and cryostages for FESEM have led to improvements in interpretable resolution to ~2 nm in “infinitely thin” biological samples (Hermann and Müller 1991; Wepf 1994; Erlandsen et al. 2000). The dimensions of many leukocyte CAMs found within the glycocalyx exceed 35–40 nm in length and therefore should be easily within the resolving power of FESEM (Barclay et al. 1997; Ushiyama et al. 1994).
A number of different CAM families are present in the glycocalyx of the human platelet, including the following: (a) a group of integrins with affinities for collagen (a2B1), laminin (a6,B1), vitronectin (a3B1), and the abundant promiscuous receptor GPIIbIIIa (CD41/CD61), which interacts with von Willibrand factor, thrombospondin, fibronectin, fibrinogen, and vitronectin; (b) leucine-rich glycoproteins, such as GPI-IX (CD42a/CD42bα,bβ); (c) two immunoglobulin-type glycoproteins, Pecam-1 and HLA class 1 β2 microglobulin; and (d) the C-type lectin family member P-selectin (CD62P) (Hsu-Lin et al. 1984; Kieffer and Phillips 1990; Lopez 1994). In an earlier study (Erlandsen et al. 1997), cryoFESEM was used to demonstrate the molecular topography of the glycocalyx in human platelets and that changes in activated platelets were consistent with the expected dimensions and shapes of known platelet CAMs. However, no specific immunolabeling of CAMs was employed.
The aim of this study was to demonstrate that use of cryomethods for preservation of cell structure together with high-resolution cryoFESEM could detect individual CAMs in the glycocalyx using a model system based on spread human platelets. Our strategy consisted of selecting distinct molecules that could be recognized by immunogold labeling. These molecules also display key structural differences resulting in unique topographical shapes. As shown in Figure 1, three major protein molecules in the platelet glycocalyx were selected for detection, i.e., the rod-shaped P-selectin molecule [CD62P; 13,000 copies per activated platelet (Berman et al. 1986)], the GPI-IX or CD42 polypeptide complex [CD42a/CD42bα,bβ; 25,000 copies per platelet (Kieffer and Phillips 1990)], and the knob-like integrin GPIIbIIIa [CD41/CD61; 50,000 copies per platelet (Kieffer and Phillips 1990)]. Using the double-layer coating method of Walther and colleagues (1995, 1997) high-resolution backscattered electron imaging was performed to demonstrate immunogold labeling by atomic number contrast and stereo imaging was used to illustrate the projection of CAMs above the surface of the platelet membrane as well as their unique topographic shapes.
Materials and Methods
Human Blood Platelets
Human blood for this study was drawn after informed consent from normal donors know to be free of any medications. As previously described (White et al. 1999), venous blood was mixed and washed with an anticoagulant (citrate/citric acid/dextrose, pH 6.5) in a ratio of 9:1 blood to anticoagulant. Washed platelets were allowed to equilibrate for 20 min at room temperature (RT) before use.
Antibodies
Mouse monoclonal antibodies against P-selectin were obtained from Dr. Rodger McEver (University of Oklahoma Health Sciences Center, Oklahoma City, OK: G1, directed against the NH2-terminal lectin domain and S12 against the short consensus repeat region) and Drs. Eugene Butcher and Aaron Warnick (Department of Pathology, Stanford University, Palo Alto, CA: WAPS12.2). Mouse MAb W5 against GPIbα and T10, a blocking antibody directed against a combined region of GPIIbIIIa, were also provided by Dr. McEver. All primary antibodies were mouse monoclonal IgG and were diluted to concentrations of 10–20 μg/ml in 10% normal sheep serum in phosphate-buffered saline, pH 7.4 (NSS–PBS). Goat anti-mouse IgG conjugated to 10-nm colloidal gold was obtained from Jackson ImmunoResearch Laboratories (West Grove, PA) and used at dilutions of 1:10–1:20 in 10% NSS–PBS.
Cell Adhesion
Platelets were allowed to spread or adhere on carbon-coated sapphire discs (2.9-mm diameter, 0.05 mm thick; Rudolf Bruegger AG, Minusio, Switzerland). To spread platelets, a drop of the platelet suspension was placed on the sapphire discs and the cells were allowed to settle for 20 min at RT. Non-attached cells were washed off in Hank's buffer. The spread cell monolayer was pre-fixed for 10 min in Hank's buffer with 0.01% glutaraldehyde, then washed in the same buffer before immunolabeling.
Antibody Labeling
The prefixed adherent platelets on the discs were incubated on the top of drops of PBS with 10% normal sheep serum (NShS–PBS) for 10 min. The discs were then blotted and transferred onto the first or primary antibody to a specific CAM. After a 30-min incubation on the first antibody, the samples were washed multiple times on drops of NShS–PBS and then transferred to a drop of immunogold marker (goat anti-mouse IgG conjugated to 10-nm colloidal gold diluted 1:20 in NShS–PBS). After a 30-min incubation period the samples were washed in PBS. All incubations were done at RT.
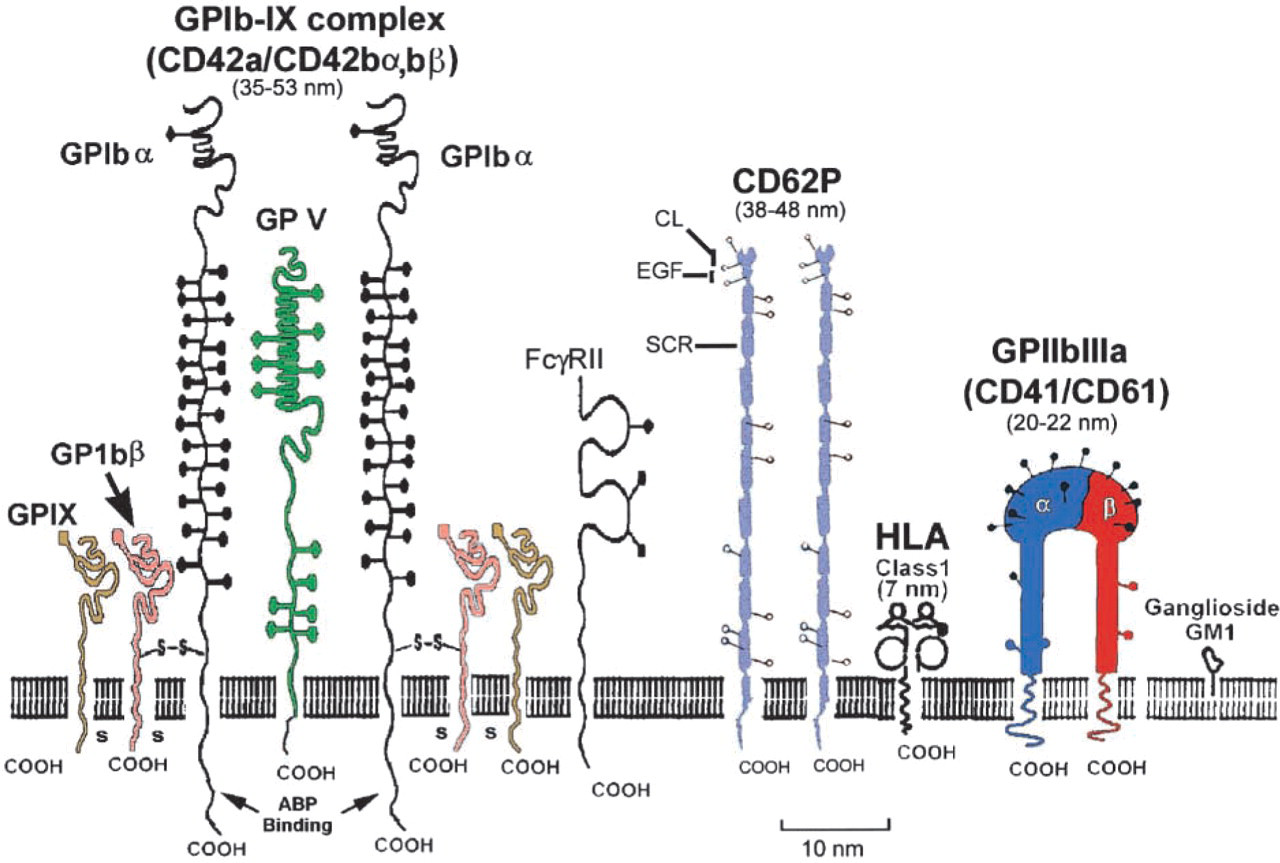
Schematic diagram of three major glycoproteins [GPI-IX complex (CD42a,/CD42bα,bβ), P-selectin (CD62P), and the integrin GPIIb-IIIa (CD41/CD61)] in the glycocalyx of human platelets. The GPI-IX complex is composed of three polypeptides (GPIbα, GPIbβ, and GPIX) that span the plasma membrane and interact with the platelet cytoskeleton via actin-binding protein (ABP). Other polypeptide chains (GPV) and the platelet Fc receptor (FcγRII) may be found in the vicinity of the GPI-IX complex. Extensive O-glycosylation (black solid circles) of GPIbα contains sialic acid and the resulting negative charge may keep the molecule extended above the surface to facilitate better accessibility of a ligand-binding domain at the NH2 terminus. The extracellular domain of GPIbα has been referred to as glycocalacin. The extracellular portion of the rod-shaped P-selectin molecule contains three domains, i.e., an amino terminal C-lectin region (CL), a short epidermal growth factor domain (EGF), and a series of nine short consensus repeats (SCR). Integrin molecules are heterodimers composed of two polypeptide chains that form one external globular head that protrudes about 20–22 nm above the platelet membrane. Dimensions shown for each glycoprotein represent length of extracellular domain (see discussion). N-linked glycosylation sites are shown by open circles for P-selectin and integrin molecules. Modified from Lopez (1994) and Barclay et al. (1997).
Post-immunolabeling Chemical Fixation
Immunolabeled spread platelet monolayers were fixed in 3% glutaraldehyde in 0.1 M Na-cacodylate buffer with 7.5% sucrose, pH 7.4, for at least 30 min. The samples were washed in the same buffer. Some samples were postfixed in 2% OsO4 in 0.1 M Na-cacodylate with 7.5% sucrose, pH 7.4, for 20 min, and then washed in the same buffer.
Sample Preparation for CryoFESEM
Immunolabeled fixed platelets were rinsed with distilled water and most of the overlying water was blotted away from the side to reduce sample thickness for better freezing results. The sapphire discs were frozen by plunging into liquid propane chilled by liquid N2 and then were cryotransferred to a modified holder for the Balzers BA-360 unit (Tech-noTrade; Manchester, NH) for double-layer coating by the method of Walther and colleagues (1995, 1997). The samples were partially freeze-dried for 45 min at –85C at a vacuum of 10−6 to 10−7 Torr. Two nm of tantalum/tungsten (Ta/W) was evaporated by electron beam deposition at an angle of 45° to the cell monolayer, followed immediately by ~7–10-nm carbon from above (90° angle). The partially freeze-dried, double-layer coated samples were cryotransferred under liquid nitrogen into a Gatan cryostage and then inserted into the Hitachi S-900 in-lens FESEM (Niessi Sangyo America; Mountain View, CA) for cryo-observation at –95C and at 10 keV accelerating voltage. High-resolution backscattered electron images were collected using a modified YAG scintillator (Autrata 1992) and recorded as tif files. Stereo images were obtained at tilt angles of ± 5°.
Measuring Height of CAMs
The height of CAMs in cryosamples was measured in high-magnification stereo pairs using the Sterecon program, a 3D, interactive, contour-based segmentation and measurement program (Erlandsen et al. 1992; Marko and Leith 1996). In viewing the stereo pair, two circles were used to measure the parallax difference between the highest and the lowest point of the macromolecule. A larger circle was placed on the membrane surface and a smaller circle was placed on the highest point of the macromolecule projecting above the membrane surface. The parallax difference (offset) between the two circles was measured in pixels and, using information on tilt angle and magnification, a height measurement in nanometers was calculated. The length of CAMs labeled indirectly with immunogold was corrected for length by subtracting 30 nm, the equivalent length for two IgG molecules.
Results
The activated spread human platelet was chosen as a model system for demonstrating individual CAMs because it possesses three unique CAMs that differ in topographic appearance (Figure 1) and because MAbs were available for all three. Unlike their normal discoid shape, human platelets fixed after spreading were seen by cryoFESEM as thin, rounded but flattened monolayers of cytoplasm (Figure 2A) with occasional pseudopods extending from the margin of the cell. High-resolution backscattered electron imaging of immunogold labeling was accomplished using atomic number contrast in which gain of the YAG detector was set so that 10-nm colloidal gold particles appeared as round white spheres. Spread fixed platelets labeled for P-selectin revealed that P-selectin molecules were distributed from edge to edge of the exposed surface of the fully spread cells (Figures 2 and 3). Immunogold-labeled P-selectin was seen as dimers or multimers of colloidal gold on rod-like extensions protruding from the surface of the platelet membrane. The vertical projection of P-selectin dimers above the membrane could clearly be seen by stereo imaging (Figure 3A) and by the length of the shadow resulting from the unidirectional shadowing of the cell at 45° with Ta/W (Figure 3B). In fortuitous backscattered-electron images at high magnification, individual molecules of P-selectin were recognizable (inset, Figure 3B). Small unlabeled knob-like protrusions were seen in the background between the labeled P-selectin CAM. In regions of the cell surface where an obstruction interfered with the angle of the metal coating, it was difficult to assess the height of immunogold-labeled P-selectin owing to lack of sufficient shadow contrast (Figure 2B).
Immunogold labeling for GPIbα in the GPI-IX complex revealed a unique pattern for immunostaining over the entire surface of the platelet unlike that seen for the rod-shaped P-selectin or the integrin GPIIbIIIa (Figures 4A-4C). Stereo analysis of the immunogold labeling for GPIbα revealed linear arrays that protruded above the platelet membrane (Figure 4C). Careful examination of the immunolabeled arrays often revealed the presence of one or two colloidal gold markers in these linear protrusions. The presence of two colloidal gold markers may represent the postulated stochiometric ratio of two GPI-IX complexes with one GPV molecule in a linear array on the surface of the membrane (Figures 1, 4A, and 4B).
The topographic appearance of the integrin GPIIbIIIa as small knob-like protrusions from the membrane surface was distinct from that of the rod-shaped P-selectin or the linear arrays seen for the GPI-IX complex. Immunogold labeling for GPIIbIIIa was detected over small knob-like projections in the dorsal surface of the fully spread platelet membrane (Figure 5). Occasionally, immunogold labeling for GPIIbIIIa was seen as dimers or multimers of immunogold particles. Careful examination of the immunolabeled membrane in Figure 5 clearly showed that not all knob-like projections were labeled with immunogold, which is consistent with the presence of other integrin molecules on the platelet membrane.
Controls for the immunogold labeling of CAM included omission of the primary antibody and the omission of the secondary antibody labeled with colloidal gold. In both cases, the end result was the absence of immunogold label on the surface molecules. In addition, the use of MAbs to CAMs having uniquely different topography showed the lack of crossreactivity among these different molecules, thus providing additional evidence for their specificity.
The projected height of the rod-shaped P-selectin molecules above the dorsal membrane of the flattened spread platelet was 48.2 ± 3.6 nm (n=4), a value similar to that reported by Ushiyama et al. (1994) for isolated molecules and multimers of P-selectin evaluated by rotary shadowing and transmission EM. Measurement of linear arrays of immunolabeled GPIbα above the membrane revealed an average height of 37.2 ± 3.3 nm (n=8), which favorably compares with the range of 35–53 nm reported by a number of investigators (Fox et al. 1988; Kieffer and Phillips 1990; Lopez 1994). The measurement of the GPIIbIIIa integrin demonstrated the greatest variation in height. The height of unlabeled integrin-like molecules (knob-shape topography) was 22.3 ± 2.7 nm (n=8), while labeled molecules were ions measured at 16.9 ± 4.2 nm (n=5). The latter value was smaller than reported values of 20–22 nm for integrin molecules (Carrell et al. 1985; Nermut et al. 1988; Barclay et al. 1997).
Discussion
The present results demonstrate that cryoFESEM possesses sufficient resolution to detect individual CAMs in the glycocalyx in a model system based on the activated and fully spread human platelet. Using immunogold labeling, three CAMs in the platelet glycocalyx were identified and confirmed as having unique topographic appearances. P-selectin (CD62P) was seen as rod-shaped protrusions above the platelet membrane, GPIbα was seen in the GPI-IX complex (CD42a/CD42bα,bβ) as part of a linear array projecting above the membrane, and GPIIbIIIa (CD41/CD61) was seen as small knoblike protrusions above the platelet membrane.
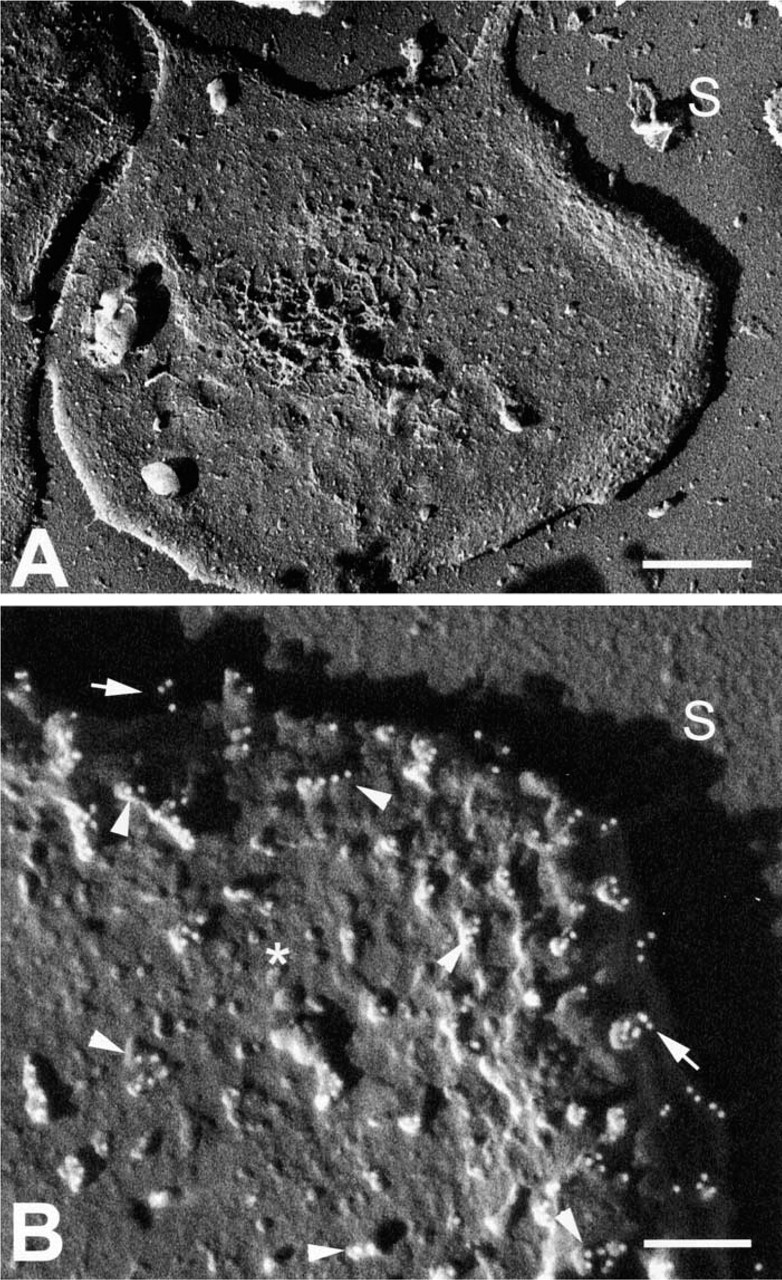
High-resolution backscattered electron images by cryo- FESEM showing a human platelet spread on a sapphire glass substrate. (
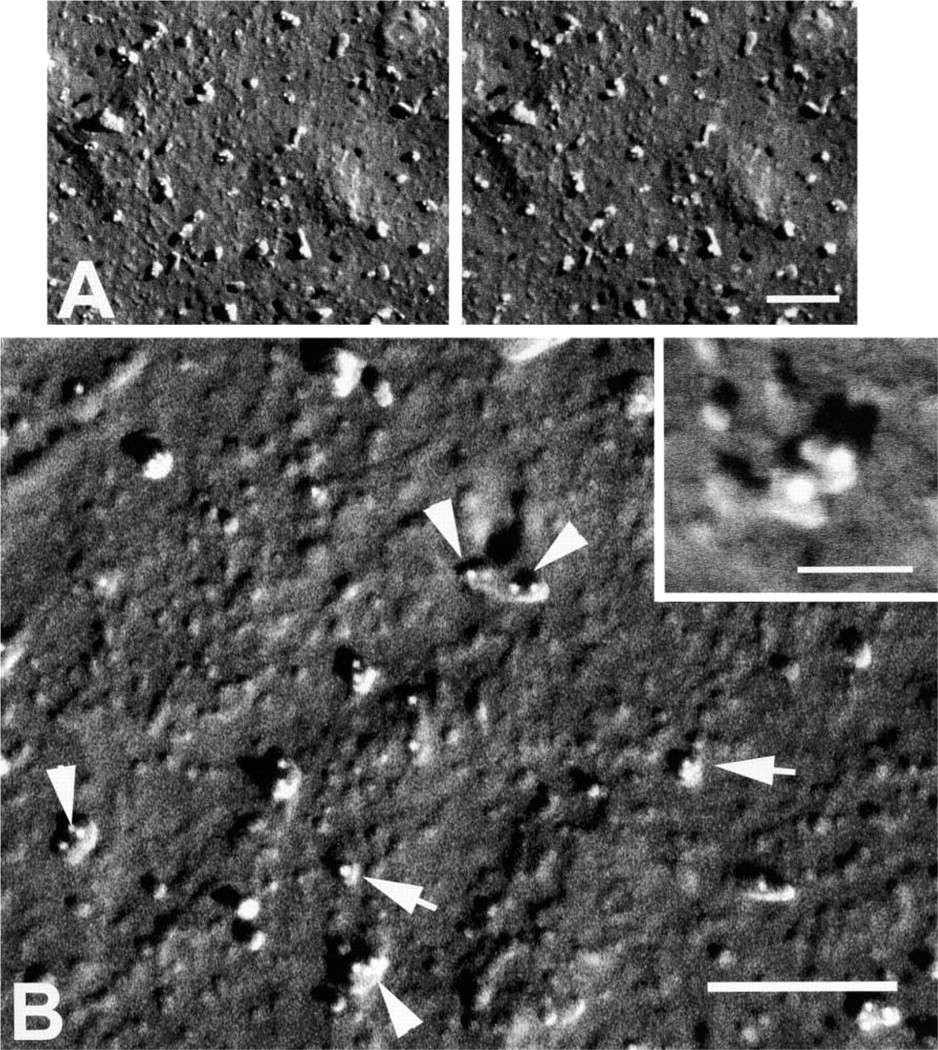
High-resolution backscattered electron cryoFESEM images of spread human platelet membranes demonstrating indirect immuno-staining for P-selectin with 10-nm colloidal gold particles (white spheres). (A) Stereo pair showing rod-like P-selectin molecules projecting above the surface of the plama membrane. Bar = 200 nm. (
Our results with immunolabeling for P-selectin support earlier work of Ushiyama et al. (1994), who investigated membrane-associated P-selectin derived from platelets and found that membrane associated P-selectin was detected as dimers or multimers. Our results with spread platelets showed that much of the P-selectin immunogold labeling also appeared as dimers or multimers on the platelet membrane, and similar results have been obtained with platelets activated in suspension (data not shown). The average length of the P-selectin molecule as measured in cryoFESEM stereo pairs using Sterecon was very close (48.2 ± 3.6 nm vs 38–48 nm) to measurements made by rotary shadowing by Ushiyama et al. (1994). However, the accuracy of the latter values should not be considered absolute values because the latter preparations were air-dried and removal of water has been shown to produce molecular collapse (Steinbrecht and Zierold 1987). Occasionally, individual P-selectin molecules were recognizable in immunolabeled dimers, suggesting that molecular definition may be possible under ideal conditions of metal shadowing and imaging (inset, Figure 3B).

High-resolution backscattered electron cryoFESEM images of the surface of human platelets indirectly immunogold-labeled for GPIbα of the GPI-IX complex. (
Lopez (1994) proposed that the GPI-IX complex would consist of the following stoichiometry: α2,β2,γ2, and δ, where α represents GPIbα, β represents GPIbβ, γ represents GPIX, and δ is GPV (Figure 1). Because GPV is associated with two GPI-IX complexes and the entire complex binds to the actin cytoskeleton, it was suggested that the physical association of GPV-(GPI-IX)2 might exist in a linear array on the cell surface (Figure 1). This interpretation was supported by our high-resolution stereo imaging of this complex (Figures 4A-4C) which demonstrated a linear array of surface projections with two gold markers associated with the complex. Linear arrays of immunogold staining for GPIbα have also been reported by Hartwig and DeSisto (1991), who examined the interrelationships of GPIbα and actin-binding protein in the platelet cytoskeleton using high-resolution transmission EM and immunogold labeling. Escolar and White (1994) examined the immunolocalization of antibody to glycocalacin (extracellular domain of GPIba) by TEM analysis of whole mounts of spread platelets. Reexamination of their images (Figure 4C in Escolar and White 1994) revealed a clustering of gold in a dimeric pattern consistent with that reported here by high-resolution cryoFESEM. The length of the GPIba chain in the GPIb-IX complex has been reported to be 60 nm, but the length of the extracellular domain or glycocalacin fragment has been calculated to be about 43 nm (Kieffer and Phillips 1990; Lopez 1994), which is very similar to the length of 37.2 ± 3.3 nm reported here as projecting above the surface. Our results show the linear arrays projecting above the surface and, together with the data of Hartwig and DeSisto (1991), provide further support for the model of Lopez (1994), which depicts the GPI-IX complex as a linear array of polypeptides attached to the cytoskeleton and protruding above the platelet membrane.
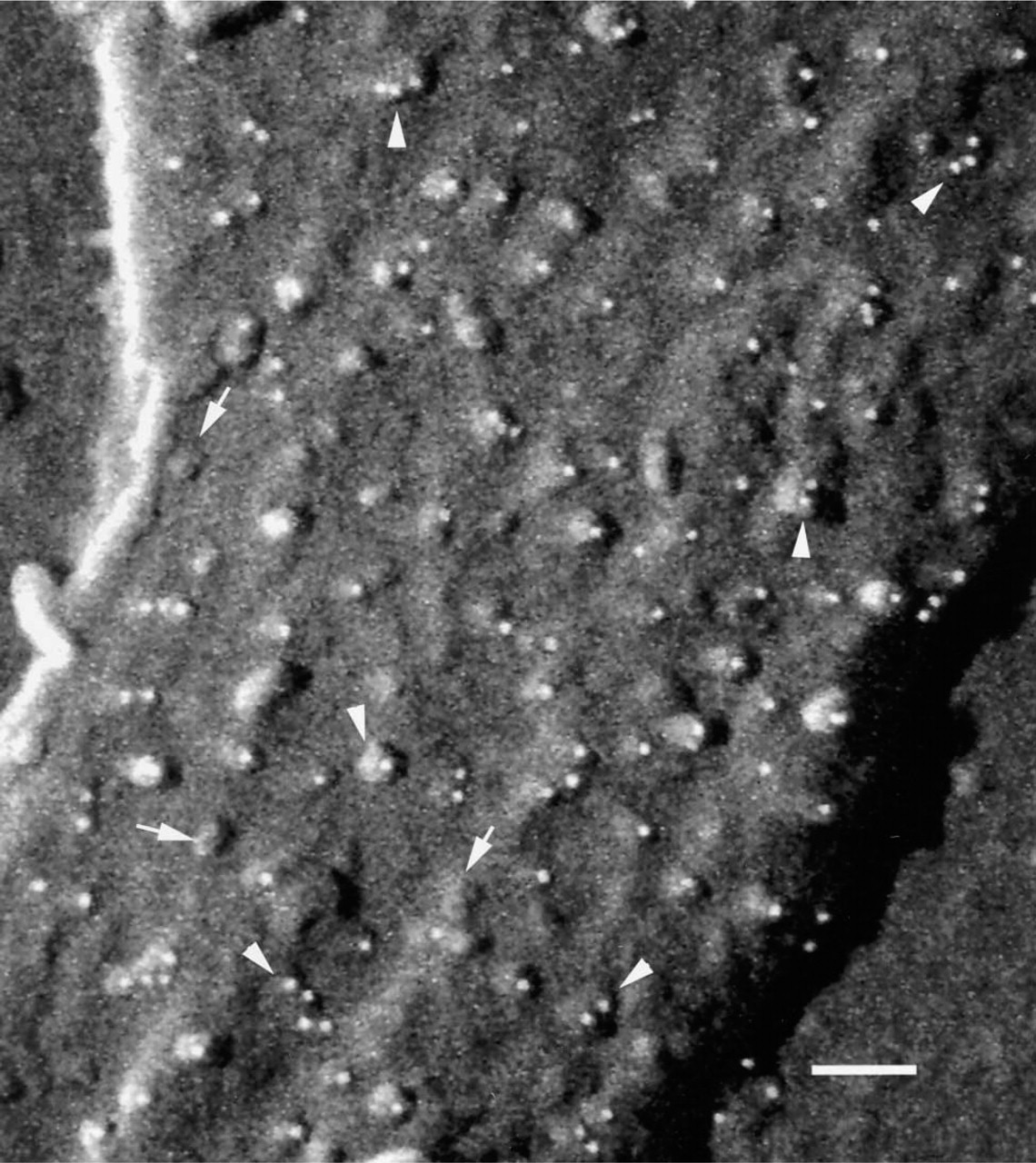
High-resolution backscattered electron cryoFESEM image of a spread human platelet showing immunogold labeling (white spheres) of the integrin, GPIIbIIIa. Integrin molecules appear as knob-like elevations above the platelet membrane. Note that the labeling for GPIIbIIIa occasionally occurs as multimers (arrowheads) and that not all integrin-like molecules (based on their topographic shape) are immunolabeled (arrows). S, substrate. Bar = 100 nm.
Comparison of CAM height measurements determined by cryoFESEM and Sterecon with values reported in the literature

aObserved values were corrected by subtracting 30 nm, the value of two IgG molecules used in the indirect immunolabeling approach.
The greatest variation in the measurement of individual CAMs was seen with the values obtained for labeled (22.3 ± 2.7 nm) and unlabeled (16.9 ± 4.2 nm) integrins. Although these are close to the reported values of 20–22 nm (Carrell et al. 1985; Nermut et al. 1988; Barclay et al. 1997), the height we measure for short CAMs tends to be underestimated because the metal shadowing needed to generate the backscattered electron signal was estimated as approximately 2 nm thick, or 10% of the reported length. Therefore, measured lengths in Sterecon for smaller CAMs may be biased towards smaller values owing to the thickness of the metal coating when lengths were determined (Table 1).
The detection and measurement of individual CAMs in the glycocalyx of human platelets have been accomplished using cryoFESEM and cryopreparative methods (Figure 6). Many factors can influence the resolution obtained in cryoFESM, including the type of specimen (“infinitely thin” vs bulky), the accelerating voltage, contamination/radiation damage, temperature, and the nature of the metal coating used to generate the secondary electron or backscattered-electron signal (Erlandsen et al. 2000). In the work reported here, high-resolution imaging was performed at magnifications greater than × 100,000 at accelerating voltages of 10 keV or greater, so image detail was carried by backscattered electrons (Type I) produced directly at the site of impact of the primary electron beam. Because the sample was coated with a thin layer of metal (~2 nm thick), the signal comes chiefly from the thickness of the metal layer and the practical limitation on resolution is the lateral diffusion of electrons which sets this limit for SEM. According to Joy (1995), with low atomic number specimens it is not unreasonable for lateral diffusion to be 7–8 nm. However, in samples coated with a thin layer of metal of high atomic number, lateral diffusion may be as small as 1–2 nm, permitting resolution on the order of 2–3 nm. This calculation is in close agreement with work by Wepf (1994) and Hermann and Müller (1991), in which analysis of cryoFESEM images from the S-phase layers of the bacterium Edinococcus radiolarens and of in vitro polymerized actin revealed a resolution of 1.5–2.0 nm. Based on the potential production of continuous metal films of approximately 0.5-nm thickness (Peters 1989), Joy (1995) has calculated that resolutions of less than 1 nm might be achievable, which could lead to improved molecular definition. Such resolution might be useful in determining the sites of glycosylation on surface molecules and in determining the presence of multimers of polypeptide chains. However, there are several challenges involved in achieving this level of resolution, including the generation of an adequate signal with extremely thin metal films, reduction in the granularity of the Cr or W coating, and the use of shallow coating angles to improve molecular definition by stereo imaging.
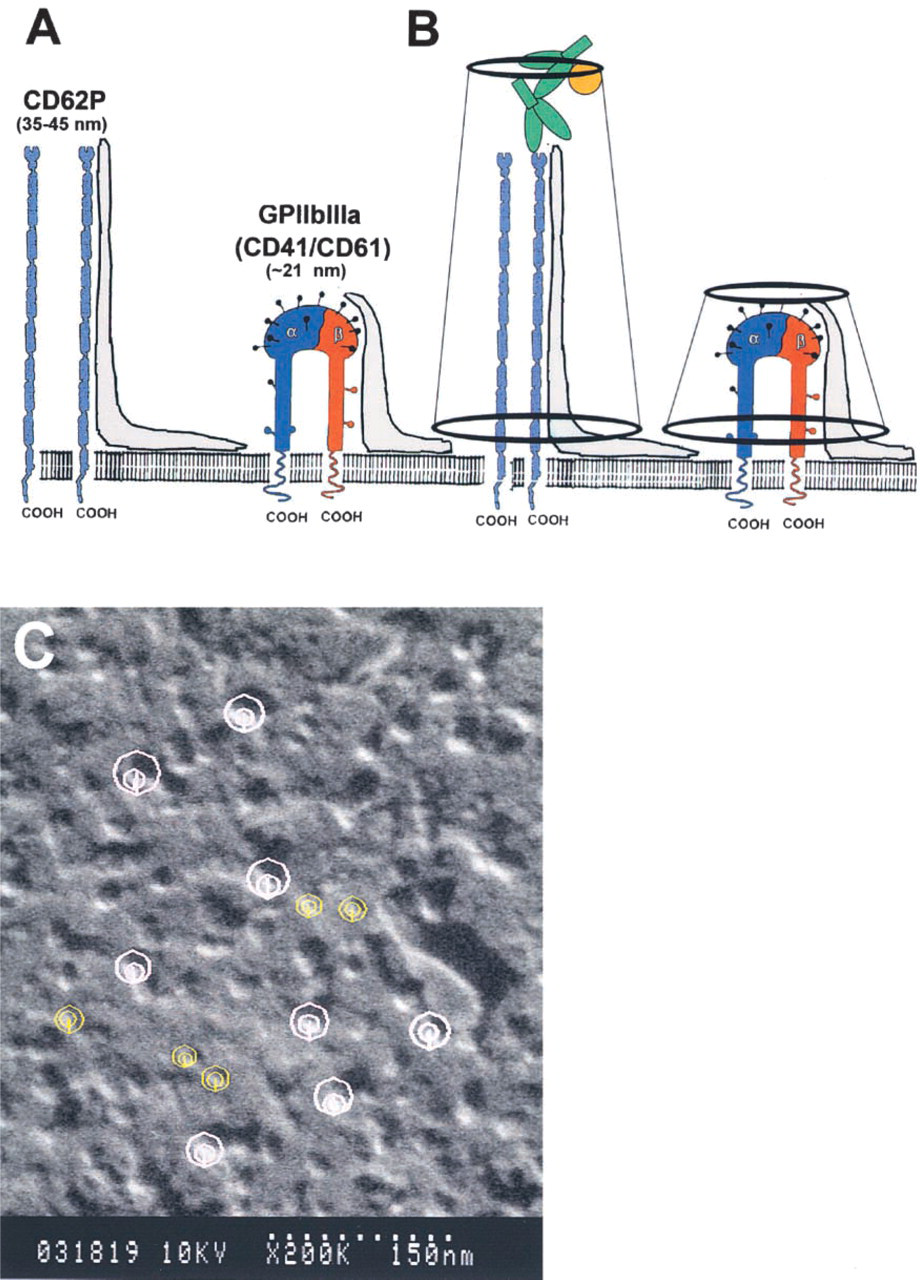
Schematic diagram and immunolabeled platelet demonstrating the method of measuring height of CAMs. (
Footnotes
Acknowledgements
Sterecon analysis work was supported by NIH NCRR grant RR01219 to the Resource for Visualization of Biological Complexity at the Wadsworth Center.
We wish to thank Drs Rodger McEver and Eugene Butcher for their generous supply of antibodies. We also thank Marcie Krumweide, Chris Frethem, and Cecile Ottenwaelter for their excellent technical assistance, and the University of Minnesota Graduate School and the Minnesota Medical Foundation for financial support.