
Abstract
Sarcoglycans are transmembrane proteins that are members of the dystrophin complex. Sarcoglycans cluster together to form a complex, which is localized in the cell membrane of skeletal, cardiac, and smooth muscle fibers. However, it is still unclear whether or not sarcoglycans are restricted to the sarcolemma. To address this issue, we examined α-, β-, δ-, and γ-sarcoglycan expression in femoral skeletal muscle from control and dystrophin-deficient mice and rats using confocal microscopy and immunoelectron microscopy. Confocal microscopy of the tissues in cross-section showed that all sarcoglycans were detected under the sarcolemma in rats and control mice. δ- and γ-sarcoglycan labeling demonstrated striations in the longitudinal section, suggesting that the proteins were expressed in the sarcoplasmic reticulum (SR) or transverse tubules (T-tubules). Moreover, such striations of both sarcoglycans were recognized in the dystrophin-deficient mouse skeletal muscle. Double labeling with phalloidin or α-actinin and δ- or γ-sarcoglycan showed different labeling patterns, indicating that δ-sarcoglycan localization was distinct from that of γ-sarcoglycan. Immunoelectron microscopy clarified that δ-sarcoglycan was localized in the terminal cisternae of the SR, while γ-sarcoglycan was found in the terminal cisternae and longitudinal SR over I-bands but not over A-bands. These data demonstrate that δ- and γ-sarcoglycans are components of the SR in skeletal muscle, suggesting that both sarcoglycans function independent of the dystrophin complex in the SR.
T
α-, β-, δ-, γ-, and ∊-sarcoglycans are all transmembrane proteins, and their respective molecular weights are about 50 kD, 43 kD, 35 kD, 35 kD, and 50 kD (Hack et al. 2000). Striated muscle contains α-, β-, δ-, and γ-sarcoglycans, while smooth muscle includes β-, δ-, and ∊-sarcoglycans (Straub et al. 1999). Structural analysis of the sarcoglycans predicts that β-, δ-, and γ-sarcoglycans are all Type II transmembrane proteins (amino terminus on the intracellular side), while α-and ∊-sarcoglycans belong to the Type I family (amino terminus on the extracellular side). β-, δ-, and γ-sarcoglycans have a cluster of cysteine residues at the carboxyl terminus, similar to receptor molecules such as epidermal growth factor (EGF) receptors and laminin (McNally et al. 1996; Chan et al. 1998). Immunoprecipitation and crosslinking examinations suggest that β-, δ-, and γ-sarcoglycans are tightly associated with one another to function as a unit, especially β- and δ-sarcoglycan (Chan et al. 1998; Hack et al. 1998). In contrast, α-sarcoglycan can be dissociated from the complex under relatively mild conditions (Chan et al. 1998). Sequence comparison between β-, δ-, and γ-sarcoglycans also reveals that they share significant homology with each other, particularly δ- and γ-sarcoglycans (Chan et al. 1998; Hack et al. 2000).
Previous reports have focused only on the sarcolemma, so little is known about whether sarcoglycans are localized in other membrane organelles such as sarcoplasmic reticulum (SR) or transverse tubules (T-tubules). Wakayama et al. (1999) first reported sarcoglycan localization at the ultrastructural level, but they did not discuss this issue. In this study, we examined sarcoglycan expression in the rat, dystrophin-deficient mdx mouse, and mouse control skeletal muscle using confocal and immunoelectron microscopy. We demonstrated that α- and β-sarcoglycans are restricted to the sarcolemma but that δ- and γ-sarcoglycans are also localized in the SR.
Materials and Methods
Animals and Tissue Preparations
All animals used in this study were cared for and handled in compliance with the Yamanashi Medical University Guidelines for the Use of Animals. We used adult female Sprague–Dawley rats, dystrophin-deficient mdx mice, and mouse controls. They were anesthetized with diethyl ether and sodium pentobarbital and perfused via the heart with 4% paraformaldehyde in 0.1 M phosphate buffer (PB, pH 7.4). The femoral muscle was removed and immersed in the fixative overnight at 4C. After rinsing in PBS, the muscle was immersed in 30% sucrose overnight at 4C. Then the muscle was embedded in OCT compound and sectioned at 10 μm thickness in a cryostat for immunohistochemistry.
Antibodies
Mouse monoclonal anti-dystrophin (NCL-DYS2), mouse monoclonal anti-β-dystroglycan (NCL-43DAG), mouse monoclonal anti-α-sarcoglycan (NCL-a-SARC), mouse monoclonal anti-β-sarcoglycan (NCL-b-SARC), mouse monoclonal anti-γ-sarcoglycan (NCL-g-SARC), and mouse monoclonal anti-δ-sarcoglycan (NCL-d-SARC) were purchased from Novocastra (Newcastle-Upon-Tyne, UK). Mouse monoclonal dystrobrevin antibody was purchased from Transduction Labs. (Lexington, KY) and goat polyclonal α-actinin antibody from Santa Cruz Labs (Santa Cruz, CA). Alexa488-conjugated phalloidin was purchased from Molecular Probes (Eugene, OR). All sarcoglycan antibodies from Novocastra Laboratories have been used in other studies and their specificity is established (Chan et al. 1998; Hack et al. 1998; Greelish et al. 1999).
Confocal Laser Scanning Microscopy
Dystrophin, dystrobrevin, and α-, β-, and δ-sarcoglycan antibodies were used at 1:10 dilution, β-dystroglycan and γ-sarcoglycan antibodies at 1:50 dilution, and α-actinin antibody at 1:100 dilution. Cryosections were treated with 1% Triton X-100 in PBS and 10% rabbit serum for 30 min each. The sections were then incubated with primary antibodies overnight at 4C, biotinylated anti-mouse Ig (Histofine ABC Kit; Nichirei, Tokyo, Japan) for 30 min at room temperature, Alexa488 coupled to streptavidin (Molecular Probes) at 1:500 dilution for 60 min, and mounted with Vectashield (Vector Labs; Burlingame, CA). For the control, PBS replaced primary antibodies. A Leica TCS-4D confocal laser scanning microscopy (Heidelberg, Germany) was used for observations with a ×63 or ×100 oil-immersion objective lens. For double labeling with phalloidin or α-actinin and δ-or γ-sarcoglycan, we used Alexa488-conjugated phalloidin at 1:200 dilution or donkey anti-goat serum (Jackson ImmunoResearch; West Grove, PA) at 1:200 dilution and biotinylated anti-mouse Ig and streptavidin-conjugated Alexa594 (Molecular Probes) at 1:500 dilution as the fluorescent agent.
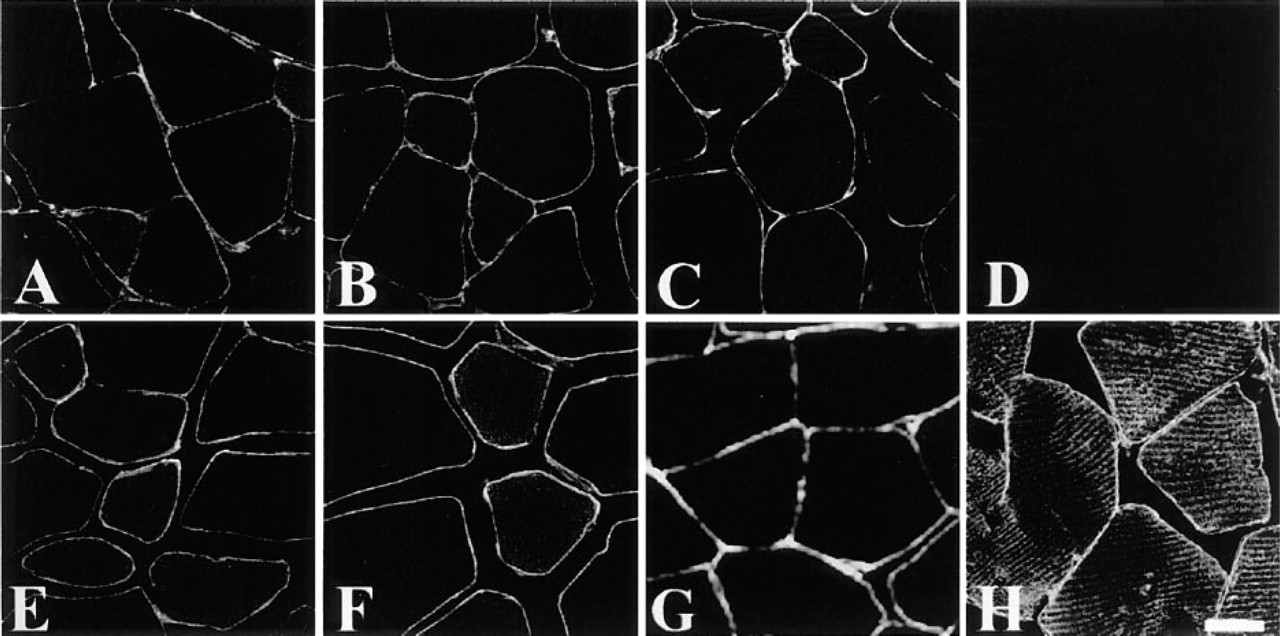
Cross-sections of rat skeletal muscle immunolabeled with dystrophin (
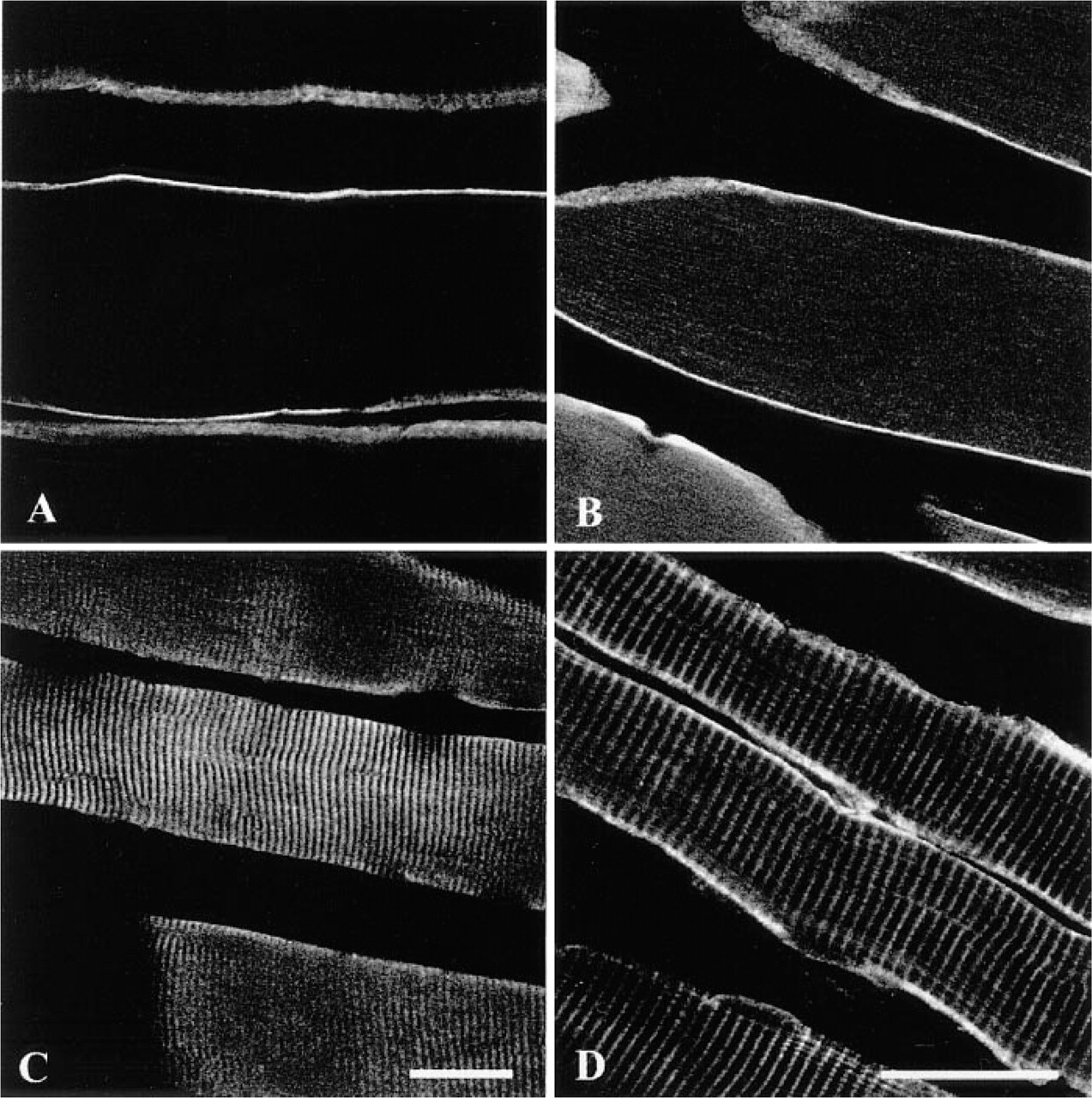
Longitudinal sections of rat skeletal muscle immunolabeled with γ-, β-, δ-, and γ-sarcoglycan antibodies (
Conventional Electron Microscopy
Conventional electron microscopic preparation was described previously (Ueda et al. 1998a). Briefly, anesthetized rats were perfused with 4% paraformaldehyde mixed with 2.5% glutaraldehyde in 0.1 M PB via the heart, and femoral skeletal muscle was immersed in the fixative overnight at 4C. After rinsing in PBS, the tissue was chopped into small pieces, treated with 1% osmium tetroxide for 60 min, dehydrated in a graded series of ethanols and acetone, and embedded in Epon (Nisshin EM; Tokyo, Japan). Ultrathin sections at 75-nm thickness were counterstained with uranyl acetate and lead citrate and observed in an electron microscope (Hitachi H-7500; Tokyo, Japan).
Immunoelectron Microscopy
Cryosections at 10-μm thickness were treated with 1% Triton X-100 and 1% H2O2 in PBS for 30 min and with 10% rabbit serum for 30 min. Then the sections were incubated with primary antibodies (anti-γ-sarcoglycan at 1:50 dilution or anti-δ-sarcoglycan at 1:10 dilution) overnight at 4C. The sections were then incubated with an ABC kit (Nichirei) according to the manufacturer's protocol, treated with a metal-enhanced DAB kit (Amersham; Poole, UK) and 1% OsO4 in PB for 60 min. The sections were dehydrated in a series of graded ethanols and embedded in Epon by the inverted gelatin capsule method. Ultrathin sections at 70 nm were stained with only uranyl acetate and were observed in an electron microscope (Hitachi H-7500).
Results
δ- or γ-sarcoglycan Expression in a Striated Pattern in the Rat and Control Mouse Skeletal Muscle
It is well known that dystrophin and dystrophin-associated proteins are expressed under the sarcolemma of the skeletal, cardiac, and smooth muscles. Our data showed that dystrophin (Figure 1A), β-dystroglycan (Figure 1B), dystrobrevin (Figure 1C), and α-, β-, δ-, and γ-sarcoglycans (Figures 1E-1H) were expressed along the sarcolemma of the rat skeletal muscle. Whereas α-sarcoglycan (Figures 1E and 2A) and β-sarcoglycan (Figures 1F and 2B) were found just beneath the sarcolemma, δ-sarcoglycan was detected not only under the sarcolemma (Figure 1G) but also in the sarcoplasm, in a striated pattern (Figure 2C). In addition, γ-sarcoglycan produced striations that appeared different from those of δ-sarcoglycan (Figure 2D). The serum control showed no immunoreactivity at all (Figure 1D). In normal mouse skeletal muscle, sarcoglycan labeling patterns were consistent with those in rat skeletal muscle (Figures 3A-3H). Highly magnified images showed that δ-sarcoglycan-labeled bands were composed of two fine striations (Figure 3G, inset). In contrast, γ-sarcoglycan showed a network-like pattern in the cross-section (Figure 3D), which appeared as thick striations in the oblique section (Figure 3H). These data suggest that δ- and γ-sarcoglycans are localized in the membrane organelles in addition to the sarcolemma in skeletal muscle.
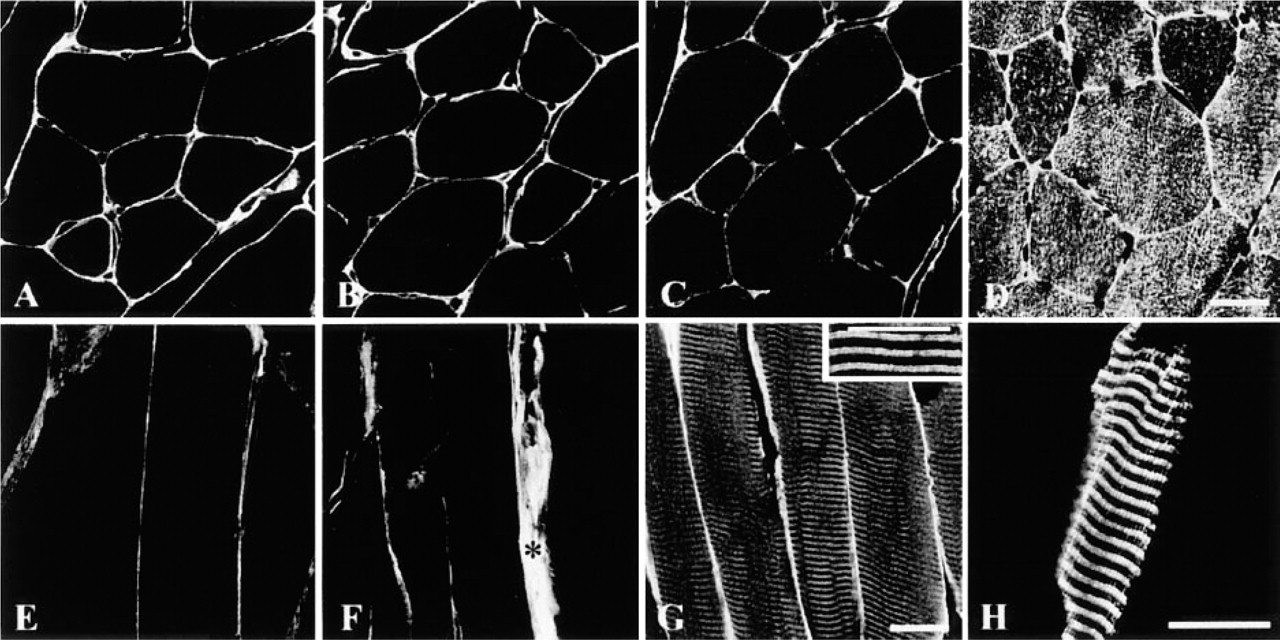
Cross- (
δ- and γ-sarcoglycan Expression in the mdx Mouse Skeletal Muscle
Previous studies reported that a primary dystrophin mutation leads to a secondary reduction or mislocalization of a number of dystrophin-associated proteins, including the sarcoglycan subunits (Ohlendieck and Campbell 1991). Strong nonspecific reaction of increased connective tissues around myofibers of the mdx mouse skeletal muscle makes it hard to determine whether or not the sarcolemma is labeled by sarcoglycan antiserum. However, γ-sarcoglycan labeling in the sarcoplasm showed a variable appearance. For example, Figure 4A shows an almost normal appearance, with regular striations like those seen in the normal mice. Perinuclear regions are strongly labeled. Figure 4B shows that regenerating muscle fibers with central nuclei (arrow) have longitudinal labeling patterns instead of striations, and some muscle fibers show a completely disorganized labeling pattern with anti-γ-sarcoglycan (Figure 4C). A sarcoplasmic banding pattern of δ-sarcoglycan is also recognized (not shown). These data suggest that sarcoglycan expression in the SR is only minimally affected by dystrophin mutations.
Longitudinal sections of mdx mouse skeletal muscle immunolabeled with γ-sarcoglycan antibody. γ-Sarcoglycan labeling (

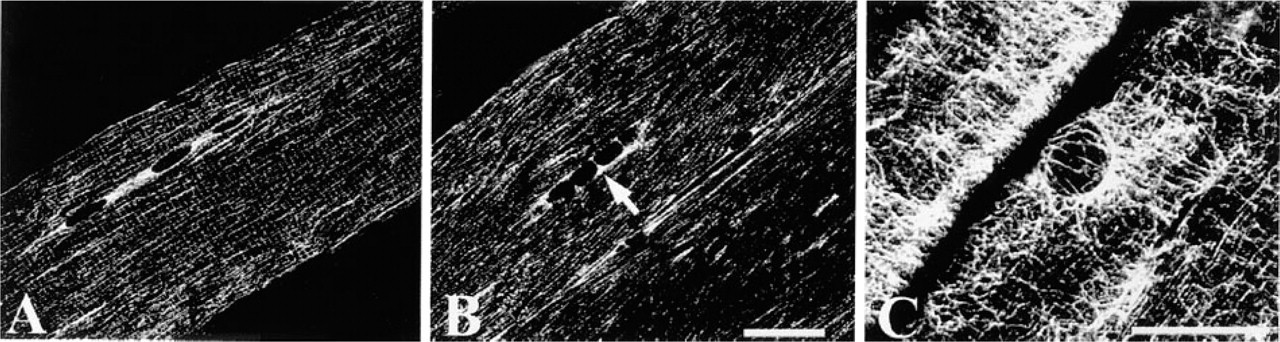
Confocal images of double labeling with phalloidin (green in
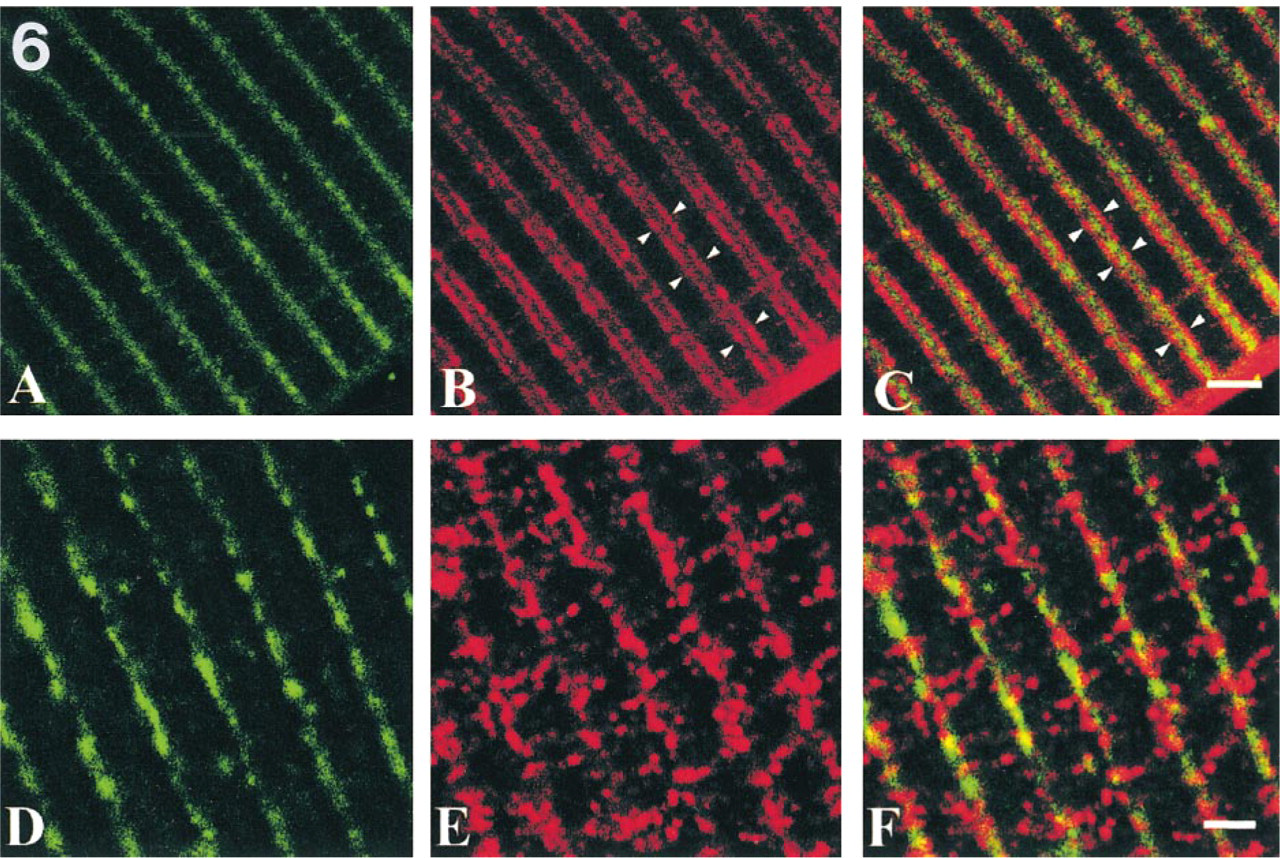
Confocal images of double labeling with α-actinin (green in
δ- and γ-sarcoglycan Topology Revealed by Double Immunolabeling with Phalloidin or α-Actinin
To determine whether or not δ- and γ-sarcoglycans are localized in the same region, we used double labeling with phalloidin or α-actinin. Phalloidin attaches to actin filaments to show I-bands, and α-actinin localizes in Z-bands in the striated muscle to anchor actin filaments (Faulkner et al. 1999). Our data showed that δ-sarcoglycan was detected in the central parts of I-bands (Figures 5A–5C) and along Z-bands (Figures 6A–6C). On the other hand, γ-sarcoglycan was found broadly over I-bands (Figures 5D–5F), especially around Z-bands (Figures 6D–6F), but was not recognized over A-bands (Figures 5D-5F). These data demonstrate that δ-sarcoglycan localization is different from that of γ-sarcoglycan.
δ- and γ-sarcoglycan Localization in the SR
To clarify the ultrastructural localization of δ- and γ-sarcoglycans, immunoelectron microscopic examinations were performed. Skeletal and cardiac muscles have special apparatuses for excitation–contraction coupling in the junctional SR (terminal cisternae) and T-tubules (Figure 7A). δ-Sarcoglycan was detected along the cell membrane of myofibers (Figure 7B, upper left inset) and in the terminal cisternae of the SR (Figure 7B and upper right inset). γ-Sarcoglycan was detected along the cell membrane (Figure 7C, upper left inset) and in the longitudinal SR over the I-band, including terminal cisternae (Figure 7C and upper right inset). These data demonstrate that δ-sarcoglycan is a component of the terminal cisternae as well as the sarcolemma, whereas γ-sarcoglycan is more broadly expressed in the SR. The schematic diagram in Figure 8 summarizes sarcoglycan localization.
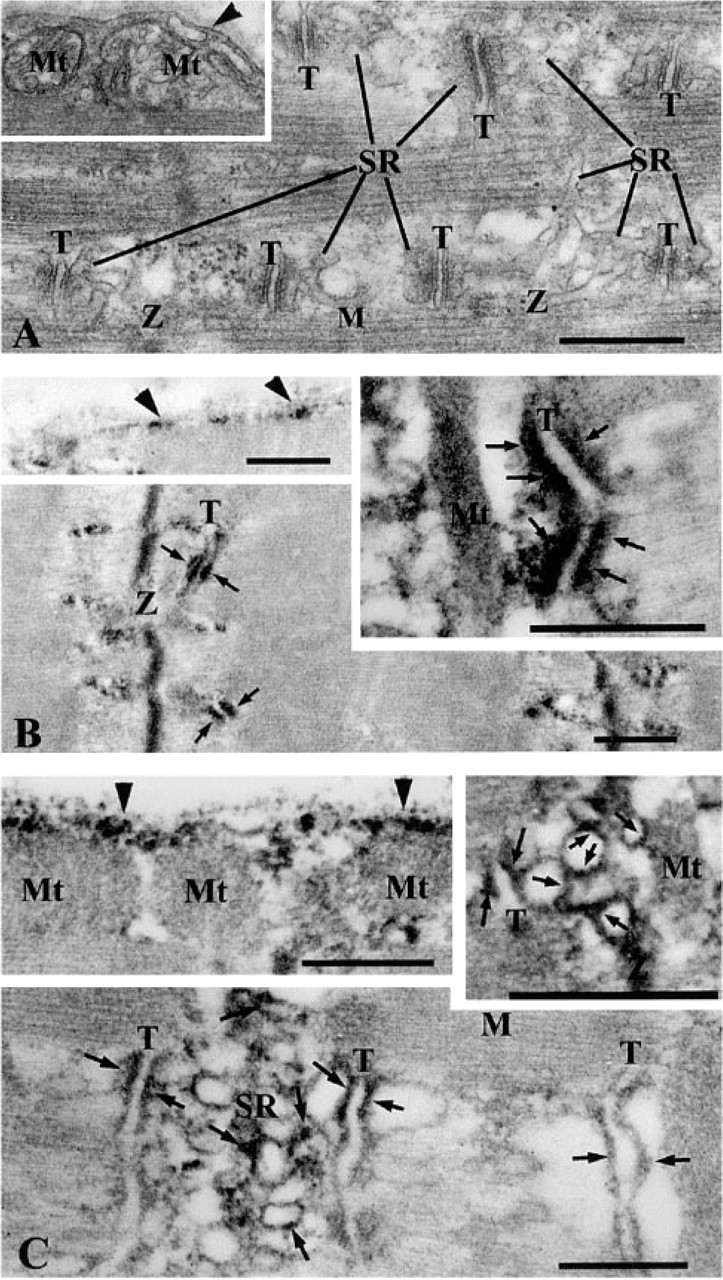
Electron micrographs of rat skeletal muscle. Conventional EMs show topology of SR, T-tubules, and myofilaments (
Discussion
In this study, we clearly demonstrated that δ-sarcoglycan was localized in the terminal cisternae of the SR, and that γ-sarcoglycan was found in the SR over I-bands, including terminal cisternae. Moreover, their expression was not affected by the absence of dystrophin. These data give a clue to sarcoglycan function.
Recent developments in molecular genetics have allowed development of α-, β-, δ-, and γ-sarcoglycan knockout mice to analyze the biological roles of these proteins (Duclos et al. 1998; Hack et al. 1998; Straub et al. 1998; Araishi et al. 1999; Durbeej et al. 2000). α-Sarcoglycan-deficient mice show ongoing muscle necrosis with age, loss of sarcolemmal integrity, and changes of absolute force (Duclos et al. 1998). β-Sarcoglycan-deficient mice exhibit progressive muscular dystrophy with extensive degeneration and regeneration, cardiomyopathy, and abnormal vascular function (Araishi et al. 1999; Durbeej et al. 2000). The BIO14.6 hamster model, which has a deletion in the δ-sarcoglycan gene (Sakamoto et al. 1997), and δ-sarcoglycan-deficient mice show muscular dystrophy and cardiomyopathy due to impairment of sarcolemmal integrity (Holt et al. 1998; Straub et al. 1998). Recently, Coral–Vazquez et al. (1999) reported that abnormal coronary vascular constriction is responsible for cardiomyopathy in sarcoglycan–sarcospan complex-disruption mice. Mice lacking γ-sarcoglycan develop a phenotype that closely parallels the human LGMD, with marked cardiomyopathy (Hack et al. 1998). To summarize these findings from sarcoglycan-deficient mice, membrane permeability changes appear to be a general feature of sarcoglycan deficiency (Hack et al. 2000). These sarcoglycan-deficient mice have in common the characteristic that the sarcoglycan complex in the sarcolemma is lost, whereas expression of dystrophin, dystroglycan, and laminin is almost intact (Duclos et al. 1998; Hack et al. 1998; Straub et al. 1998; Araishi et al. 1999; Durbeej et al. 2000). Duclos et al. (1998) reported that α-dystroglycan was enriched and fully glycosylated but was not tightly associated with membranes in α-sarcoglycan-deficient mice. In addition, Hack et al. (1998) reported that γ-sarcoglycan deficiency in mice produced an almost complete secondary reduction of both β- and δ-sarcoglycans but that their mRNA was normally produced. They also found that staining for dystrophin, β-dystroglycan, and laminin was intact, suggesting that the mechanical dystrophin–dystroglycan–laminin link was unaffected by γ-sarcoglycan deficiency (Hack et al. 1998, 1999). In fact, the present data on mdx mice show that δ- and γ-sarcoglycan expression in the SR appears to be independent of dystrophin deficiency.
Terminal cisternae are important structures for calcium release–calcium uptake mechanisms through triad configuration with a T-tubule. It is assumed that calcium influx through dihydropiridine receptors on the T-tubule induces calcium release from the terminal cisternae via ryanodine receptors, resulting in muscle contraction. The SR contains proteins that are characteristically associated with calcium regulation, such as ryanodine receptors, calsequestrin, sarcoplasmic or endoplasmic reticulum calcium ATPase (SERCA), phospholamban, triadin, junctin, and myotonic dystrophy protein kinase (Jorgensen et al. 1983; Knudson et al. 1993; Jones et al. 1995; Franzini–Armstrong and Protasi 1997; Shimokawa et al. 1997; Ueda et al. 1998b). Sequence analysis reveals that β-, δ-, and γ-sarcoglycans contain cysteine residues at the carboxyl terminus (extracellular part; Chan et al. 1998), and cysteine-rich motifs are commonly found in many proteins, such as laminin or EGF, that serve as receptors (McNally et al. 1996). Accordingly, it is possible that the β-, δ-, and γ-subunits also function as receptors for a yet unidentified ligand (Chan et al. 1998). Ueyama et al. (1998) found that the BIO14.6 hamster has abnormal ryanodine receptor expression in the cardiac muscle, suggesting abnormal calcium regulation caused by δ-sarcoglycan deficiency. Taken together with our present data demonstrating δ-sarcoglycan localization in the junctional SR, we suggest that δ-sarcoglycan may be associated with calcium regulation. On the other hand, the γ-sarcoglycan localization is quite unusual. To our knowledge, no protein is reported to be localized in the SR over I-bands. Two possible functions of γ-sarcoglycan are hypothesized. First, γ-sarcoglycan may be related to calcium metabolism. The cytoplasmic domain of γ-sarcoglycan has five tyrosine residues, and recent studies imply that bidirectional signaling with integrins may involve the sarcoglycan subunits (Yoshida et al. 1998). Second, γ-sarcoglycan may anchor the SR to myofilaments or Z-bands during muscle contraction and relaxation. Hack et al. (1999) suggested that both δ- and γ-sarcoglycan have critical and non-redundant roles for muscle function. Although the present findings appear to support this idea, further studies are needed to address these hypotheses. In conclusion, the present study demonstrates that δ- and γ-sarcoglycans are SR proteins, and that fact sheds light on the functions of sarcoglycans.
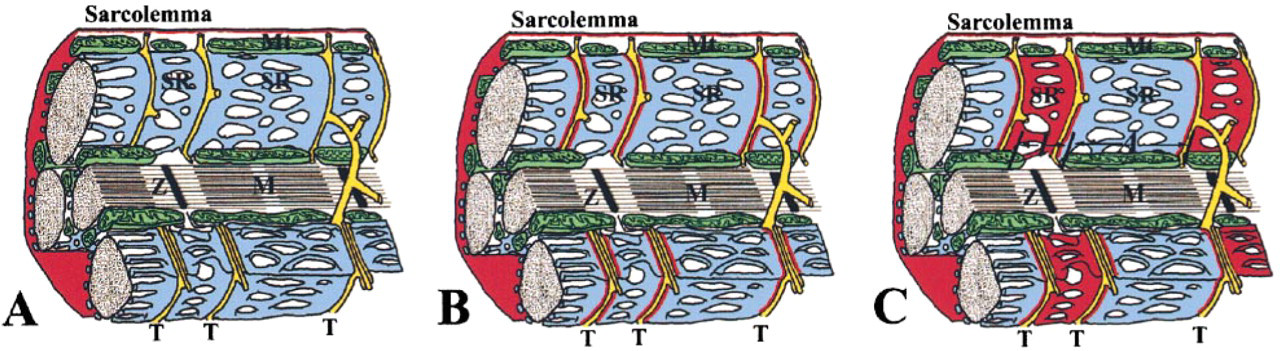
Schematic diagram of localization of α-, β-, δ-, and γ-sarcoglycan (in red). α- and β-sarcoglycans are restricted to the sarcolemma (
Footnotes
Acknowledgements
Supported in part by grants from the Ichiro Kanehara Foundation and the TV YAMANASHI Science Development Fund.