
Abstract
Epoxy resins provide optimal tissue morphology at both the light and the electron microscopic level and therefore enable correlative studies on semithin and thin sections from the same tissue block. Here we report on an approach to retain these advantages for immunolabeling studies by adapting and combining well-known techniques, i.e., surface etching with sodium ethoxide and heat-mediated antigen retrieval. We propose a simple procedure for immunostaining semithin and thin epoxy resin sections. To check its applicability, well characterized, commercially available antibodies (against E-cadherin, α-catenin, and β-catenin) were used on sections of human small intestine. By light microscopy, the immunostaining efficiency was compared on cryo-, paraffin, and epoxy semithin sections processed in parallel. The most detailed results were obtained on semithin sections, where the labeling precisely delineated the lateral plasma membrane of the enterocytes. At the electron microscopic level the procedure did not damage the structures and allowed an efficient, reproducible immunogold labeling extending homogeneously over exceptionally wide tissue areas. The three antibodies specifically labeled the zonula adherens of the junctional complex between epithelial cells and, in agreement with light microscopic observations, the lateral plasma membrane.
T
Materials and Methods
Tissue Preparation
Samples of human proximal jejunum were surgically removed from three patients (informed consent was obtained in accordance with the Helsinki Declaration of 1975) from the edges of the anastomosing sites during the preparation of a Roux-en-Y loop (Nagel 1991). Immediately after excision, the specimens were fixed in a solution of 4% formaldehyde freshly prepared from paraformaldehyde according to Karnovsky (1965) in PBS, pH 7.4, minced into small blocks, transferred into fresh fixative solutions, and stored for 12–18 hr at 4C. After washing in PBS, the tissue blocks destined for cryosectioning were soaked overnight in 20% sucrose (freeze protection), enclosed in OCT Tissue Tek compound (Miles; Elkhart, IN), and frozen in isopentane cooled by liquid nitrogen. The specimens were stored at −80C until sectioning. The other tissue blocks were dehydrated in ascending concentrations of ethanol and either embedded in the paraffin-equivalent Histocomp (Vogel; Giessen, Germany) or in the epoxy resin Agar 100 (Serva; Heidelberg, Germany).
Primary Antibodies and Specificity
Monoclonal primary antibodies generated in mice (Transduction Laboratories; Lexington, KY) were used. They were directed against the C-terminal domains of E-cadherin (clone 36, catalogue no. C20820), α-catenin (clone 5, catalogue no. C21620), and β-catenin (clone 14, catalogue no. C19220). The specificity of the antibodies was checked as recommended by the manufacturer by immunoblotting of lysates provided together with the antibodies: A 431 cells for E-cadherin, human endothelial cells for α-catenin, and HeLa cells for β-catenin. The polypeptides in the lysates were separated by SDS-PAGE using a 9–19% gradient polyacrylamide gel and electrophoretically transferred to a Protran nitrocellulose membrane (Schleicher & Schuell; Dassel, Germany). The membrane was blocked with 5% skimmed milk and incubated for 60 min with the primary antibodies diluted according to the instructions of the manufacturer. For detection, the blots were incubated for 60 min with peroxidase-conjugated goat anti-mouse IgG (Cappel; Organon Teknika, West Chester, PA), developed with the chemiluminescent reagent NEN (Nalgene; Boston, MA) and exposed on Hyperfilm (Amersham Life Science; Little Chalfont, UK). The antibodies recognized bands at 120 kD (anti-E-cadherin), 102 kD (anti-α-catenin), and 92 kD (anti-β-catenin) (Figure 1) in agreement with published data.
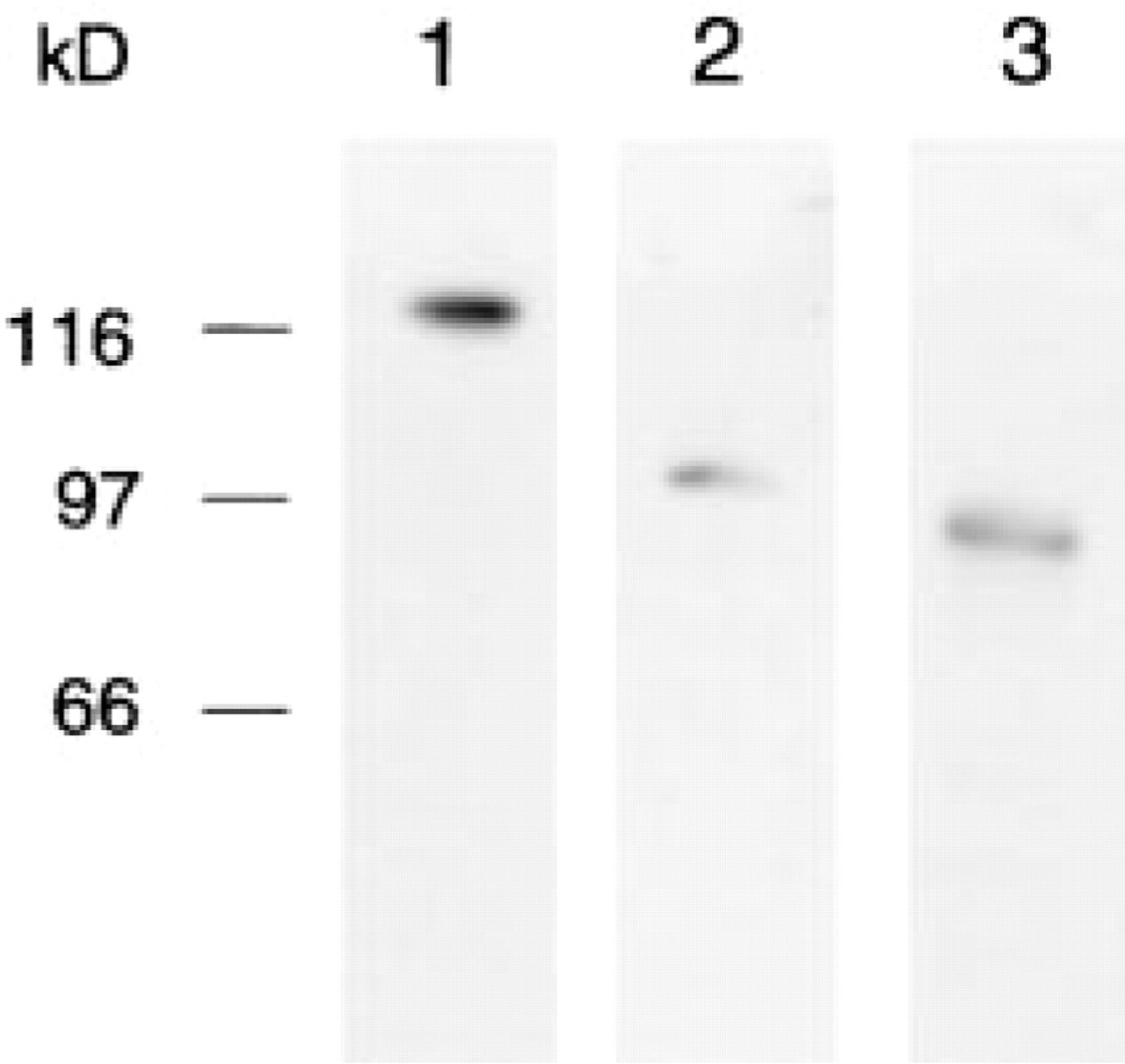
Specificity of the antibodies shown by immunoblotting. A lysate of A 431 cells immunoblotted with anti-E-cadherin labels a band at about 120 kD (Lane 1). Lysate of human endothelial cells immunoblotted with anti-α-catenin labels a band at about 102 kD (Lane 2). Extract of HeLa cells immunoblotted with anti-β-catenin labels a band at about 92 kD (Lane 3). Molecular weight standards are indicated at left.
Labeling Reagents
For immunohistochemistry, a biotin-conjugated affinitypurified goat anti-mouse IgG (Jackson ImmunoResearch Laboratories; West Grove, PA) was used as secondary antibody. For cryosections, a dichlorotriazinyl–fluorescein (DTAF)-labeled streptavidin (Jackson ImmunoResearch) was applied, while for both paraffin and epoxy resin sections peroxidase-labeled streptavidin (Jackson ImmunoResearch) was used. In thin sections, the immunoreaction was visualized with a goat anti-mouse IgG+IgM antibody conjugated to 10-nm gold particles (British BioCell; Cardiff, UK). All incubation steps were carried out in a humid chamber.
Immunohistochemical Procedures
Cryosections (8 μm thick) of frozen samples were cut with a cryostat Leica CM 1900 (Leica; Bensheim, Germany) at −20C, collected on SuperFrost Plus microscope slides (Menzel-Gläser; Braunschweig, Germany), air-dried, and rehydrated. Because formaldehyde-fixed tissue was used, in some cases antigen retrieval was performed by transferring the cryosections for 30 min into 0.01 M citrate buffer, pH 6.0, previously heated in a microwave oven to 95C.
After washing in PBS and blocking nonspecific protein binding with 5% BSA in PBS for 30 min at room temperature (RT), the sections were incubated overnight at 4C with the primary antibodies as indicated in Table 1. The sections were washed in PBS, treated with the biotin-labeled secondary antibody (5 μg/ml) for 60 min (RT) and then with streptavidin labeled with the fluorochrome DTAF (5 μg/ml) for 30 min at RT. After mounting with Mowiol (Sigma; Deisenhofen, Germany), the sections were examined with a Leitz Dialux 22 microscope equipped with fluorescence. Photomicrographs were taken on Kodak TMAX 400 film (Eastman Kodak; Rochester, NY).
Dilutions of the monoclonal primary antibodies a
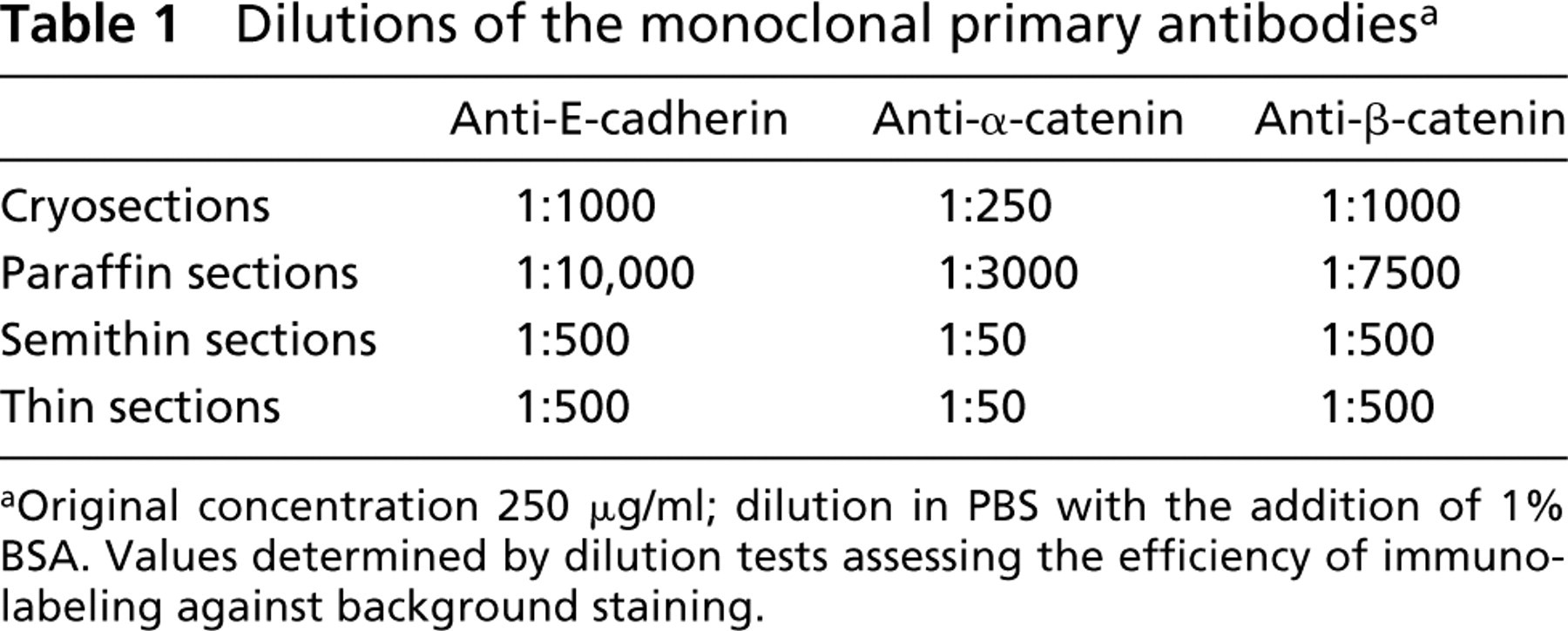
aOriginal concentration 250 μg/ml; dilution in PBS with the addition of 1% BSA. Values determined by dilution tests assessing the efficiency of immunolabeling against background staining.
Paraffin sections (about 4 μm thick) were cut and collected on glass slides pretreated with 3-(triethoxysilyl)propylamine (silane; Merck, Darmstadt, Germany). The sections were dewaxed in xylene and rehydrated in descending concentrations of ethanol. Endogenous peroxidase activity was blocked by treating the sections with 0.6% H2O2 in 96% ethanol for 20 minutes at RT. Later, these sections were processed for antigen retrieval and immunostaining in the same way as the epoxy resin semithin sections (see below).
For epoxy resin sections, semithin sections (about 1 μm thick) were cut with a Leica Ultracut R ultramicrotome, collected on silane-treated slides, and air-dried at RT. A controlled corrosion of the section surface was obtained by etching the sections for at least 15 min (shorter etching times were insufficient) in a solution composed of saturated sodium hydroxide in absolute ethanol (sodium ethoxide) diluted to 50% with absolute ethanol and subsequently rehydrating in descending concentrations of ethanol. After rehydration, for both paraffin and epoxy sections antigen retrieval was performed in a microwave oven (Sharp R-2S67; Sharp, Hamburg, Germany) by heating the sections in 0.01 M citrate buffer, pH 6.0 (Cattoretti et al. 1992), at 700 W three times for 5 min and then allowing them to cool for 30 min. Antigen retrieval greatly improved the immunostaining on paraffin sections and was essential for epoxy sections because etching alone did not allow antigen detection.
Nonspecific protein binding was blocked by treating the sections with 5% BSA dissolved in PBS for 30 min at RT. After a short rinse with PBS, the sections were incubated overnight at 4C with the primary antibodies diluted as indicated in Table 1, washed in PBS, treated for 60 min with biotinylated goat anti-mouse IgG (5 μg/ml), and then with peroxidase-labeled streptavidin (5 μg/ml) for 30 min, both at RT. Peroxidase labeling was visualized with 3,3′-diaminobenzidine (Sigma) as chromogen. The sections were washed in PBS and subsequently in distilled water, dehydrated in ascending concentrations of ethanol, cleared in xylene, mounted in Entellan (Merck, Darmstadt, Germany) and examined with a Leica DMLB microscope.
Thin sections (about 70 nm thick) were prepared from the same block used for LM immunohistochemistry and collected on nickel grids. During the immunostaining procedure, incubation steps were carried out by floating the sections on 20-μl drops of each solution; washing steps were performed by dipping the grids into the washing solution. Epoxy resin was partially corroded by etching the sections for 10 sec in saturated sodium ethoxide diluted to 50% with absolute ethanol. Etching times of 5, 10, 15, 20, and 25 seconds had been tested in advance (see Results). After rehydration and washing in distilled water, antigen retrieval was carried out by heating the sections for 10 min to 95C and then cooling them to 21C at a rate of 0.04C/sec in a thermocycler UNO-Thermoblock (Biometra; Göttingen, Germany). In preliminary tests, times of 5, 10, and 15 min for antigen retrieval had been checked. After washing in three changes of PBS, nonspecific protein binding was blocked by incubation with 5% BSA in PBS for 30 min at RT. Incubation with the various primary antibodies (for working concentrations see Table 1) was carried out overnight at 4C. After application of the 10-nm gold-labeled secondary antibody for 60 min at RT at a concentration of 0.8 μg/ml and subsequent washing in PBS, the immunoreaction was stabilized with 2.5% glutaraldehyde in PBS for 10 min (Merighi 1992). The sections were counterstained with uranyl acetate and lead citrate and examined in an electron microscope (Zeiss EM 10 CR) at an acceleration voltage of 80 kV.
Control Experiments
Control sections were treated in the same way as the experimental sections, but either omitting the primary antibodies or replacing them with normal mouse serum in the same concentrations as the applied antibodies.
Results
Light Microscopy
After application of antibodies against E-cadherin, low magnification of cryo-, paraffin, and epoxy sections showed immunostaining of the epithelium (Figures 2a–2c). The staining was diffuse in cryosections, but in paraffin and epoxy sections it was localized to the lateral plasma membrane of the enterocytes throughout the crypt–villous axis. Around the goblet cells the staining was more pronounced (Figures 2a–2c).
Although cryosections did not require pretreatment for detection of E-cadherin and catenins, their immersion in citrate buffer previously heated in a microwave oven improved the pattern of the fluorescent staining, which appeared more confined to the plasma membranes and in places had a dotted appearance (Figures 2a and 2d).
To achieve homogeneous and reliable staining on paraffin sections, heating in a microwave oven was necessary. Epoxy sections were not labeled without a sequential pretreatment of surface corrosion (etching) with sodium ethoxide and microwave heating. In both paraffin (Figures 2b and 2e) and epoxy sections (Figures 2c and 2f) the immunolabeling was always restricted to the plasma membrane of the epithelial cells and had a dotted appearance. This aspect was particularly evident in epoxy sections (Figures 2f and 3a). In cryosections, the very intense fluorescence at the crypt base prevented clear immunostaining of single cells (Figure 2a). In contrast, in paraffin (Figure 2b) and even more so in epoxy sections (Figures 2c and 3a), the Paneth cells were morphologically recognizable because of their granules and a pronounced specific labeling of the lateral plasma membrane, whereas the basal plasma membrane remained unlabeled (Figures 2b, 2c and 3a). This peculiar staining of Paneth cells was especially evident with Nomarski interference optics (Figure 3a) and was confirmed for all three antibodies applied (data not shown).
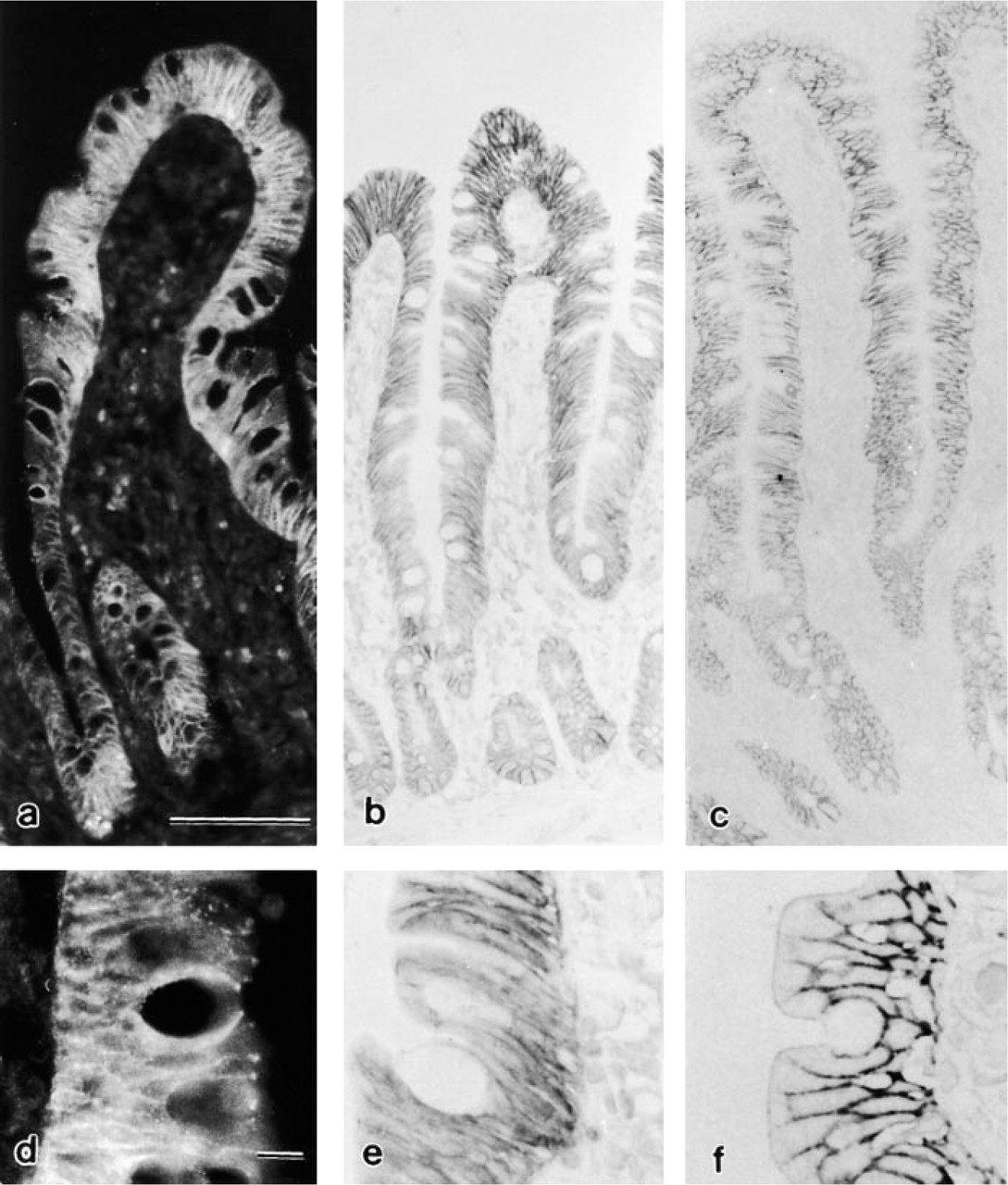
Immunolocalization of E-cadherin in the epithelium of the human small intestine. (
Electron Microscopy
To obtain optimal labeling without loss of specificity, it was necessary to evaluate etching time, different concentrations of the etching reagent, and various heating periods for antigen retrieval. Thin sections that were etched for 5 sec in sodium ethoxide diluted to 50% or for 20 sec in a dilution of 25% and subsequently heated showed good morphological preservation but only poor labeling. In contrast, if the sections were subjected to 15, 20, or 25 sec of etching in 50% solution, labeling improved but the long etching periods caused serious damage to the sections, such as the formation of holes, the size of which increased during heating. In extreme cases the sections were reduced to small remnants close to the bars of the grid. Concerning the heating time a threshold of 10 min was set, the labeling being insufficient below this time and not enhanced by prolonged exposure. Therefore, in our approach the best and most reliable results were obtained by etching the thin sections for 10 sec in 50% sodium ethoxide and subsequently heating them to 95C for 10 min, followed by a gradual, controlled decrease of temperature to 21C (see Materials and Methods). This procedure did not damage the morphology and allowed analysis at the EM level of the immunolabeling throughout the whole section over an area equivalent to that of a semithin section, as shown in Figures 3a (LM) and 3b (EM) at the same magnification. A survey of cells and/or tissue areas could be obtained and specific structures could be examined by high magnification using the same (Figures 3c and 3d) or adjacent sections (Figure 4e). Alternatively, immunolabeling first detected at high magnification (Figure 3d) could be traced back via intermediate (Figure 3c) to survey magnification (Figure 3b) to establish the exact position of the labeled epithelial cells along the crypt–villous axis.
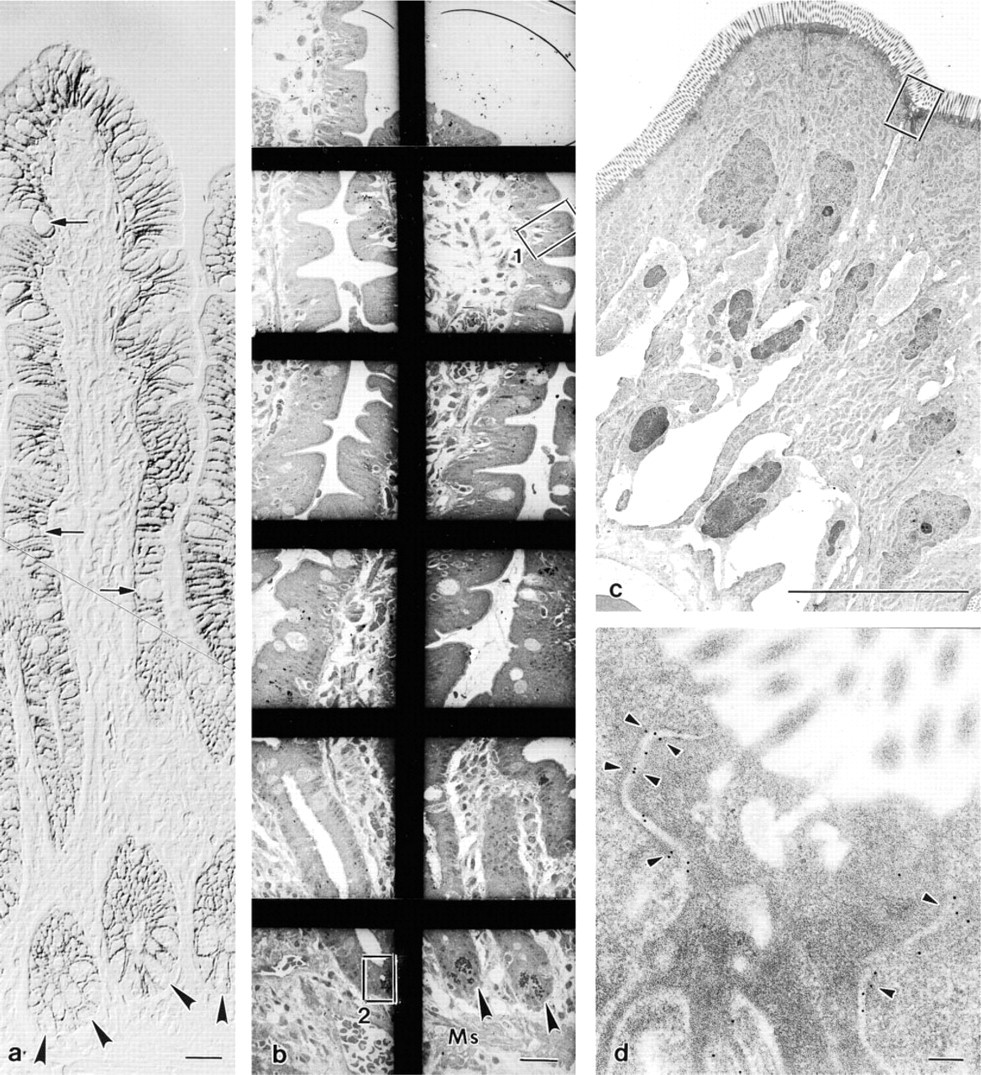
(
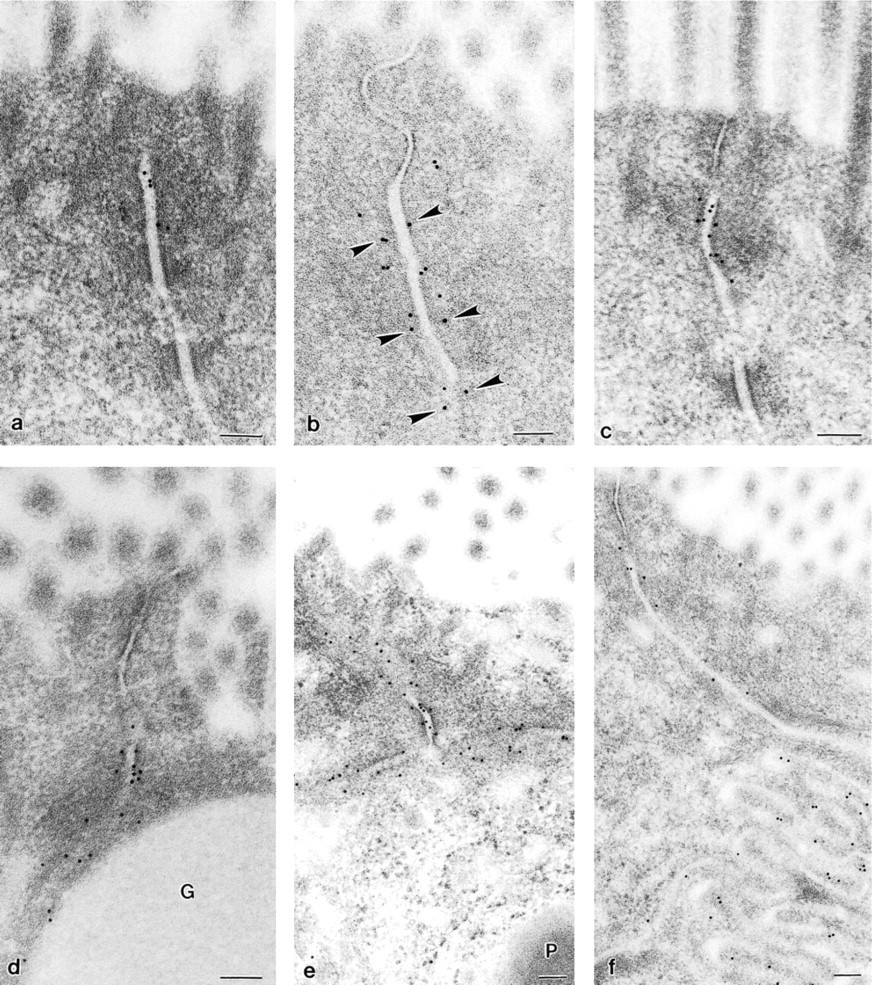
The junctional complex between enterocytes (
In human small intestine, all three antibodies showed immunoreactions at the level of the junctional complex between enterocytes, where the gold particles preferentially bordered the cytoplasmic sides of the zonula adherens but left free both zonula occludens and desmosomes (Figures 4a and 4c). However, some particles appeared dislocated inside the intercellular space (Figures 4a and 4c). A bilateral correspondence of the gold labeling could occasionally be observed between two enterocyte plasma membranes (Figure 4b). Immunostaining was always pronounced at the junctions between enterocytes and goblet cells (Figure 4d) and especially between two adjacent Paneth cells (Figure 4e). Furthermore, in agreement with LM, the immunolabeling was localized with varying density below the junctional complex along the lateral plasma membrane of all epithelial cells (villus and crypt; Figures 4d and 4f). Reliable and specific labeling was obtained with all three antibodies. However, the lowest density of labeling was seen with the antibody against E-cadherin (compare Figure 4a with Figures 4b and 4c).
Discussion
Advantages Over Other Procedures of Epoxy Sections for Immunostaining
Once a method to remove the embedding medium from epoxy semithin sections was found (Mayor et al. 1961), conventional stains for LM directly correlating between semithin and consecutive thin sections were applied (Lane and Europa 1965), and later enzyme histochemistry and immunohistochemistry also became applicable at both LM and EM levels (reviewed in Moriarty 1973). Nevertheless, the use of epoxy sections for immunostaining remained sporadic, especially at the EM level, probably because of the difficulty in exactly evaluating the effects produced on the embedding medium by various etching agents (Horobin and Proctor 1982). Successful EM immunostaining, as demonstrated here, requires some experimenting to determine the right balance between the concentration of the etching agent and etching time (a) to provide sufficient permeability of the surface of thin sections for antibody access while avoiding structural damage and (b) to unmask antigens hidden by covalent bonds formed between epoxy resin and biological material during polymerization (Kellenberger et al. 1987). To circumvent this time-consuming screening, a radical treatment of the sections has been proposed, consisting of deplasticization (i.e., complete removal of plastic medium, Epon, or Araldite) of semithin and thin sections, which after immunostaining were dehydrated and re-embedded for further sectioning and direct examination in EM, respectively (Lackie et al. 1985; Mar et al. 1987; Mar and Wight 1988; Brorson and Skjörton 1995).
Acrylic resins such as Lowicryl, LR White, and LR Gold are preferable for several reasons. They are water-compatible and therefore require only a partial dehydration of the tissue, thus improving its reactivity with antibodies. Second, the hydrophilic properties of the plastic medium reduce background staining and make these resins especially suited for postembedding immunocytochemistry (Newman and Hobot 1987). A further characteristic is a weak adherence between acrylic resins and biological material during polymerization (reviewed in Newman and Hobot 1993) which, for example in the case of Lowicryl, leads to a roughness of the thin section surface with a penetrable relief calculated at 3–6 nm (Kellenberger et al. 1987). However, this facilitated penetration does not warrant efficient labeling of Lowicryl sections if an adequate concentration of randomly dispersed proteins is not present in the tissue, so that sufficient epitopes do not become available within the penetrable surface layer of the section (Kellenberger et al. 1987).
LR White and LR Gold might have other technical drawbacks, such as tissue extraction and shrinkage caused by its exposure to the damaging influence of the plastic monomer during the embedding procedure, depending on the temperature conditions under which polymerization occurred (Newman 1987).
Cryo-ultrathin sections, already used for enzyme histochemistry in the 1960s (Leduc et al. 1967) have been proposed for immunocytochemistry (Tokuyasu 1980). In recent years they became the method of choice because no pretreatment of the sections is required for antigen–antibody binding, as thawed cryosections are fully penetrable (Tokuyasu 1980). However, cryo-ultramicrotomy presents more difficulties than anticipated. First, if fixation with aldehydes is applied (and it is used in most cases; see Griffiths 1993), the cytoplasm turns into a gel-like mass with irregular penetrability for both primary and secondary antibodies. If fixation is limited or omitted, an almost continuous loss of soluble material occurs as soon as the section is immersed in aqueous solutions during the labeling process. Second, each tissue block can be cut only once. The ultracryosections are small because they are prepared from frozen specimens a priori small and additionally trimmed to select the tissue region of interest. Nevertheless, serial ultrathin cryosections can be used for the application of different antibodies to localize the corresponding antigens in the same tissue region in successive sections (Griffiths 1993). This, however, is achieved just as well on epoxy-embedded tissue treated as described here. In addition, there are the following advantages: each tissue block can be repeatedly cut for both LM and EM immunolabeling; the etching followed by heating allows the epitopes to be unmasked and structures preserved in thin sections large enough to maintain topographic relationships between cells distributed over wide tissue areas. In our special case, the entire height of the human jejunal mucosa (about 850 μm in our specimens) could be explored and immunolabeling of cells with peculiar locations, e.g., Paneth cells, could be compared with other kinds of cells farther away on the same section.
Antigen retrieval is a section pretreatment that exposes hidden epitopes (Shi et al. 1997). As discussed above, the embedding medium can induce masking of the epitopes and restriction of antibody access. In addition, the aldehydes used as fixatives significantly influence antigen detection. As known for a long time (Fraenkel–Conrat and Olcott 1948), they crosslink proteins, resulting in a masking effect that must be reversed. Therefore, antigen retrieval is often a prerequisite to performing an immunohistochemical investigation, and the so-called microwave procedure (Shi et al. 1991) is particularly suitable. In our approach, antigen retrieval together with preceding etching was an essential pretreatment for both semithin and thin sections to detect E-cadherin and its associated proteins in a constant and reproducible manner.
Localization of E-cadherin and Catenins
E-cadherin, α-catenin, and β-catenin belong to a family of adhesion molecules that are expressed at the junctions of the adherens type (for review see Yap et al. 1997). The zonula adherens represents a special kind of junction and is located at the junctional complex sealing epithelial cells apically (Farquhar and Palade 1963). Hence, intestinal epithelium represents an ideal tissue in which to test the presence of adhesion molecules.
In agreement with previous investigations (Hermiston and Gordon 1995; Miyashiro et al. 1995; Haftek et al. 1996; Inomata et al. 1996; Senda et al. 1996; Mahmoud et al. 1997; Valizadeh et al. 1997; Takahashi et al. 1998), our results at the LM level show that E-cadherin, α-catenin, and β-catenin are localized along the lateral plasma membrane of the enterocytes. As shown by others (Miyashiro et al. 1995; Senda et al. 1996), the use of cryosections occasionally leads to a slight cytoplasmic staining which, however, is not confirmed in paraffin or semithin sections. This staining might be artificially caused by the diffusion of the detection product or might be due to the sum of the labeled plasma membranes within the section thickness. Paraffin sections have an advantage over cryosections because structural preservation is improved. Epoxy semithin sections have not been used previously to localize E-cadherin and its undercoat proteins α- and β-catenin. In our study they provide the best morphological tissue preservation, together with efficient and reliable immunostaining. Semithin sections can be prepared that allow a survey of the spatial distribution of the antigen with the option to study its subcellular expression on consecutive thin sections.
In thin epoxy sections of human small intestine, E-cadherin and/or its associated proteins α- and β-catenin are distinctly localized at the zonula adherens. The tight junction zone and the desmosomes are free of immunogold particles. Our findings are in agreement with electron microscopic investigations of different segments of animal and human intestine, using either acrylic resin sections (Miyashiro et al. 1995; Haftek et al. 1996; Senda et al. 1996) or ultrathin cryosections (Itoh et al. 1993). Although epoxy embedding media are mostly referred to as less suitable for postembedding immunoelectron microscopy, the density of gold grains achieved in our specimens is not inferior to that shown in previous investigations. Furthermore, we were able to detect immunolabeling along the lateral plasma membrane below the junctional complex according to the LM immunostaining, not shown before at the EM level.
The results of this study demonstrate that epoxy sections from the same tissue block are suitable for performing immunostaining at both the light and electron microscopic level by maintaining all advantages these media have: best tissue preservation, good infiltration qualities, block samples easy to cut, excellent structure contrast, and stability under the electron beam.
Footnotes
Acknowledgements
We wish to thank Prof Dr G. Hünefeld (Göttingen–Weende) for help in collecting the specimens, Prof Dr E. Ungewickell for critical reading of the manuscript, Ms C. Lichtenberg for technical assistance, Ms. A. Hundt for photographic assistance, and Ms S. Fryk for linguistic correction of the manuscript.