
Abstract
Pre-embedding double immunogold–silver labeling using two ultrasmall gold conjugates has not been attempted previously because a means of distinguishing labels by conjugates of identical sizes was lacking. This study investigated the feasibility of creating a particle size segregation between two ultrasmall gold conjugates through sequential immunogold incubations and silver enhancements. Two primary antibodies, mouse anti-synaptophysin and rabbit anti-glial fibrillary acidic protein (GFAP), were used in the model system. Differentiation of the double labeling was achieved by incubating with one ultrasmall gold conjugate, followed by silver enhancement, and then incubating with the second ultrasmall gold conjugate, followed by additional silver enhancement. This resulted in two groups of silver-enhanced particles: smaller particles enhanced once and larger particles enhanced twice. Electron microscopic examination revealed two readily distinguished populations of gold–silver particles within the appropriate structures, with very little size overlap. The quality of the ultrastructure permitted identification of most subcellular organelles. This procedure provides for the first time a pre-embedding immunogold–silver labeling protocol that allows the precise subcellular co-localization of multiple antigens.
I
Ultrasmall gold (≤ 1.0-nm) conjugates introduced by Leunissen (reviewed by van de Plas and Leunissen 1993) provided a new marker system that has greater labeling sensitivity and better penetration than conventional gold particle conjugates, and higher spatial resolution than enzyme conjugates. These advantages, along with the development of silver enhancement reagents, have made it possible to apply pre-embedding immunogold–silver techniques to label intracellular antigens, including proteins associated with microtubules (Gutekunst et al. 1995), ribosomes (Feng et al. 1997), endoplasmic reticulum (Gilmor et al. 1996), and nuclear aggregates (Gutekunst et al. 1998), and even to localize specific intracellular and extracellular epitopes of intrinsic membrane proteins (Hersch et al. 1997).
Ultrasmall gold conjugates and silver enhancement have also been widely employed for the co-localization of multiple antigens, although only in conjunction with conventional gold conjugates (Verkade et al. 1997) or enzyme conjugates (Rouse et al. 2000). In practice, if ultrasmall gold conjugates were exclusively used in multiple labeling, silver enhancement would be required to produce particles with a different shape or size to distinguish labeling by each conjugate. We considered that it might be possible to achieve this goal through the use of sequential immunogold labeling and silver enhancement. Thus, the first ultrasmall gold conjugates would be enhanced twice and would therefore generate a size different from the second ultrasmall gold conjugate, which would be enhanced only once (Figure 1). We report here a new procedure that successfully applies this concept to pre-embedding double immunogold–silver labeling using two ultrasmall gold conjugates of different species with a commercially available silver enhancement reagent.
Materials and Methods
Antibodies and Reagents
Primary antibodies, rabbit anti-glial fibrillary acidic protein (GFAP; Dako, Carpinteria, CA) and mouse anti-synaptophysin (Chemicon International; Temecula, CA), were chosen because of their known specific antigen localization in the central nervous system. The mutually exclusive distributions of their respective antigens also made these two antibodies ideal for testing the specificity of this procedure and evaluating particle size segregation.
All secondary antibodies (ultrasmall gold conjugated F(ab′)2 fragment of goat anti-rabbit IgG and goat anti-mouse IgG), acetylated bovine serum albumin (BSA-c), bovine serum albumin (BSA), coldwater fish skin gelatin (CWFS gelatin), and R-gent SE-EM electron microscopy grade silver enhancement reagent were purchased from Aurion (Wageningen, The Netherlands). Enhancement Conditioning Solution (ECS) prototype was provided by Aurion.
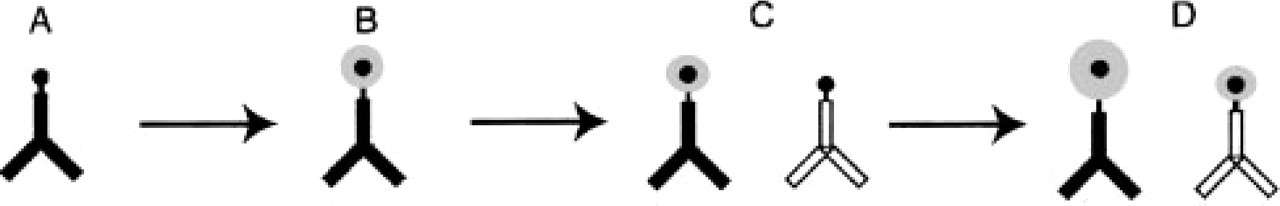
Schematic illustration of the procedure for pre-embedding double immunogold labeling with silver enhancement. (
Tissue Preparation
All procedures involving animals were approved by the Emory University Animal Care and Use Committee. Adult C57B16 mice were anesthetized with chloral hydrate (400 mg/kg), and perfused transcardially with 150 ml of 3% freshly depolymerized paraformaldehyde and 0.15% glutaraldehyde in 0.1 M phosphate buffer (PB; pH 7.2–7.4) at a flow rate of 10 ml/min. Brains were removed and further fixed in the same fixative for 1 hr at 4C and sectioned coronally at 50 μm using a Vibratome (Technical Products International; St Louis, MO). The sections were then collected and washed thoroughly with PB.
Double Immunogold Labeling
All steps and durations for double immunogold labeling and silver enhancement are listed in Table 1. In general, washed sections were placed in PB containing 0.1% sodium borohydride to inactivate residual aldehyde groups in the tissue sections. Sections were then washed with PB several times until the solution was clear of bubbles. To improve reagent penetration, the sections were then treated with PB containing 0.05% Triton X-100. To prevent nonspecific “sticking” of the immunoreagents, sections were incubated in blocking solution, which was PBS, pH 7.4, containing 5% normal goat serum (NGS), 5% BSA, and 0.1% CWFS gelatin. After blocking, sections were incubated in a mixture of rabbit anti-GFAP (1:4000) and mouse anti-synaptophysin (1:4000) primary antibodies diluted with PBS containing 0.2% BSA-c (PBS/BSA-c, pH 7.4). After washes with PBS/BSA-c, sections were incubated in the first secondary antibody conjugate, which was ultrasmall gold-conjugated F(ab′)2 fragments of goat anti-mouse IgG diluted 1:100 with PBS/BSA-c. To remove unbound secondary antibody, sections were washed thoroughly with PBS/BSA-c and then with PB. After washes, sections were prepared for the silver enhancement (see below).
Protocol for pre-embedding double immunogold–silver labeling

After the first silver enhancement, sections were washed with PB and then incubated with the second secondary antibody conjugate, ultrasmall gold-conjugated F(ab′)2 fragments of goat anti-rabbit IgG diluted 1:100 in PBS/BSA-c. Sections were washed with PBS/BSA-c and then PB. Before the second silver enhancement, sections were fixed with 2.5% glutaraldehyde in PB to crosslink immunoreagents in the tissue to prevent the loss of labeling during subsequent processing. All immuno-incubations were done with gentle agitation at 4C.
For each double labeling of GFAP and synaptophysin, three additional groups of sections were processed in parallel as controls. They were incubated with either rabbit anti-GFAP, mouse anti-synaptophysin, or buffer solution only. After the primary antibody incubations, all four groups were treated identically.
Silver Enhancement
Sections were washed with ECS. Sections were then transferred to R-gent SE-EM silver enhancement solution in a clean 24-well culture plate shielded from bright light and incubated at room temperature for 90 min for the first silver enhancement and 60 min for the second. The R-gent SE-EM silver enhancement solution was prepared according to the manufacturer's instructions. The enhancement was stopped by placing sections in ECS containing 0.03 M sodium thiosulfate and then further washing with ECS.
Tissue Processing for Electron Microscopy
Sections were washed with PB, fixed with 0.5% osmium tetroxide for 15 min, dehydrated, and flat-embedded in Eponate 12 resin (Ted Pella; Redding, CA) between two sheets of Aclar film (Electron Microscopy Sciences; Fort Washington, PA). After resin polymerization, small pieces of flat-embedded sections were dissected from the dorsal striatum, mounted on plastic stubs, and sectioned en face. Silver colored ultrathin sections were stained with 4% aqueous uranyl acetate and lead citrate (Reynolds 1963).
Particle Size Analysis
Immunogold-labeled axon terminals and glial processes in ultrathin sections from the surface of the Vibratome sections were photographed at ×30,000 with a Hitachi H-7500 transmission electron microscope. Negatives were digitized, and the particle diameters were measured using NIH Image software (Rasband; National Institute of Health, Bethesda, MD). For the measurement of synaptophysin-associated immunogold–silver particles, complete terminals were chosen and a total of 450 particles within axon terminals were measured. For the measurement of GFAP-associated immunogold–silver particles, an area of 0.05 μm2 was selected from several glial process profiles, and a total of 511 particles within glial process were measured. For each group of particles, diameter mean and SEM were calculated and particle diameters were plotted in a frequency distribution. A two-tailed Student's t-test was also conducted to evaluate particle size segregation between two groups.
Results and Discussion
This study aimed to develop a procedure for the application of two ultrasmall gold conjugates for the co-localization of two antigens. In the present protocol, two ultrasmall gold conjugates were applied sequentially to label synaptophysin and GFAP, and each was followed by a silver enhancement step. The first gold conjugate (to synaptophysin) was enhanced once alone for 90 min and once again when the second gold conjugate (to GFAP) was enhanced for 60 min. TEM examination of the labeling revealed two distinct populations of particles based on sizes (Figure 2). Small GFAP gold particles were associated primarily with filamentous structures in the perikarya and processes of glial cells, and only rarely were large particles seen within glial profiles. Large synaptophysin gold particles were predominantly present in axon terminals, where they were associated with synaptic vesicles. Occasionally, small particles were seen in axon terminals mixed in with the larger particles. In control samples from which one of the primary antibodies had been omitted, there were no gold-silver particles of relevant size concentrated within the expected structures, except for a few randomly distributed background particles.
Particle Size Segregation
The size of the particles (mean ± SEM) within glial processes was 11.2 ± 0.14 nm and in axon terminals was 36.3 ± 0.36 nm. This difference was highly significant by a two-tailed Student's t-test (p < 0.001). When the size frequency distributions of particles found in glial processes and axon terminals were plotted on the same graph, a very small overlap was evident (Figure 3). This overlap was mainly due to a few small particles in axon terminals. The number of particles in axon terminals whose size overlapped with particles in glia represented 2.9% of all particles in axon terminals. The region on the frequency distribution curve for particles in axon terminals that overlapped with particles in glia was more than two standard deviations below the mean.
Evidently, homogeneity of silver enhancement is crucial for creating the particle size segregation in this procedure. With any enhancement reagent, a certain degree of variability is expected (Burry et al. 1992; Rufner et al. 1995). Considering this variability and the particle size suitability for our application, we sought a greater difference in size than is normally used in on-grid double immunogold labeling with conventional gold conjugates. In theory, the larger the size difference is between the two groups of particles, the smaller the degree of overlap. However, as a practical matter, particles must be large enough to ensure adequate visualization but not so large as to obscure the underlying ultrastructure.
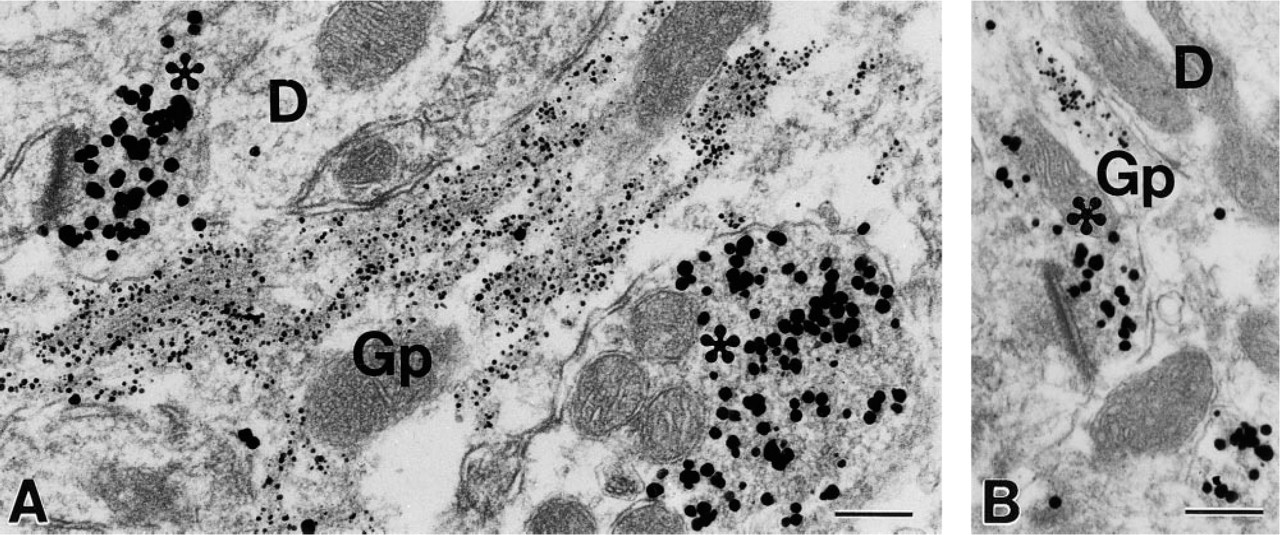
Electron micrographs (
It should be pointed out that the particle sizes and the amount of segregation produced here are not fixed parameters. On the basis of our experience, it is possible to generate combinations of smaller particles, which may be advantageous for co-localizing more contiguous antigens. In this experiment, the enhancement duration required for obtaining a final particle size of 10–12 nm was first determined through a series of single labeling experiments (data not shown) and then applied to the second silver enhancement. The duration of the first silver enhancement was then systematically varied to create particle size segregation.
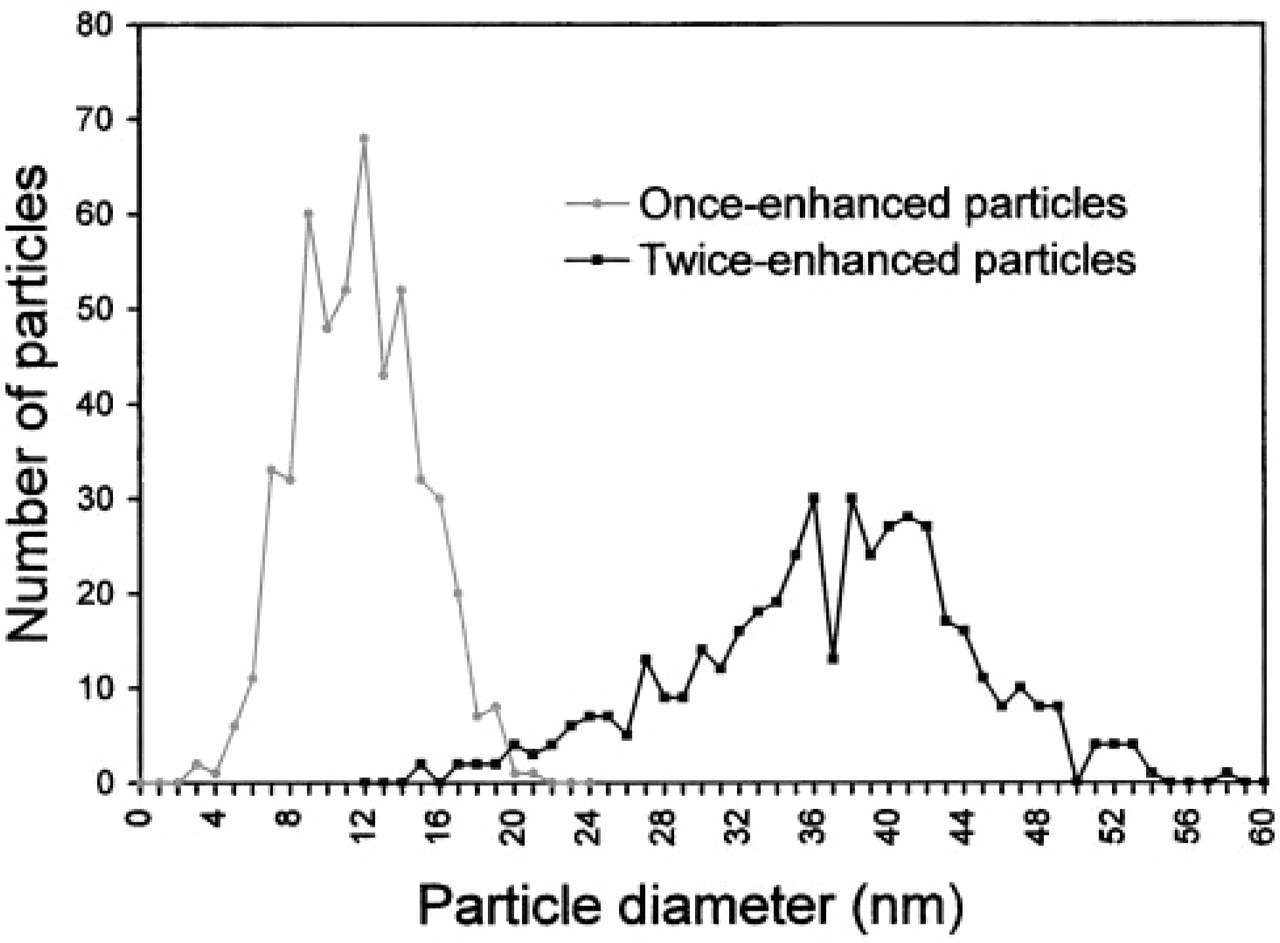
Comparison of immunogold-silver particle size distribution, showing little overlap. The gray line represents once-enhanced particles within glial profiles (n = 511; mean ± SEM = 11.2 ±0.14 nm). The black line shows the twice-enhanced particles within axon terminals (n = 450; mean ± SEM = 36.3 ± 0.36 nm).
Ultrastructural Quality
Adequate preservation of ultrastructure has been a major obstacle in pre-embedding immunogold labeling. In most immunogold–silver labeling protocols, silver enhancement is carried out after postfixation with glutaraldehyde and before postfixation with osmium tetroxide, at which point the ultrastructure is still vulnerable. However, many silver enhancement reagents require extensive water washes before and after the enhancement, which is the major cause of deterioration in ultrastructural quality. In sequential pre-embedding double immunogold–silver labeling, preserving tissue ultrastructure is even more challenging because additional fixation with a high concentration of glutaraldehyde cannot be applied before the first enhancement due to its possible effect on subsequent antibody–antigen recognition. In search of an alternative to the water washes, we experimented with several solutions. The commonly used phosphate buffer or saline solution is not suitable because both silver phosphate and silver chloride form insoluble precipitates. Reportedly, citrate buffer and HEPES buffer have been applied with success (Danscher 1981; Burry et al. 1992). In our experiments, a manufacturer-supplied Enhancement Conditioning Solution (ECS; see Materials and Methods) was used as a washing solution to remove phosphate and chloride ions before silver enhancement. Sodium thiosulfate was added to the ECS and used as a washing solution to eliminate residual silver ions after silver enhancement. Figure 2 shows examples of the ultrastructure obtained from our experimental samples. Membrane outline was fairly intact, and all major organelles were clearly recognizable.
Other Possibilities
In this procedure, we chose two primary antibodies from different species. However, it has been reported that secondary antibodies on the surface of gold particles can be inactivated by silver enhancement (Bienz et al. 1986) and thus become unable to bind to their antigen. This feature may enable the use of two primary antibodies of the same species in pre-embedding double immunogold labeling.
Footnotes
Acknowledgements
Supported by NIH R01-NS35255 (CAG, SMH), NIH P01 HD35576 (GMS, SMH), and NSF IBN-9983078 (CAG).
We wish to thank Dr Lorin Freedman and Dr Howard Rees for editorial assistance. We acknowledge the Office of the Dean for Research, Emory University School of Medicine, for supporting the Neurology Microscopy Core Laboratory where this project was conducted.