
Abstract
Keywords
P
The morphological development and ultrastructural alterations of peroxisomes during ontogenesis have been studied in various organs (Keller et al. 1993), including liver (Tsukada et al. 1968; Essner 1969; Stefanini et al. 1985; Espeel et al. 1990, 1993, 1997), kidney (Goeckermann and Vigil 1975; Pipan and Psenicnik 1975; Stefanini et al. 1994; Oberley et al. 1995; Johkura et al. 1998), intestine (Pipan and Psenicnik 1975; Calvert and Menard 1978; Dauça et al. 1996), lung (Schneeberger 1972), and brain (Houdou et al. 1991; Itoh et al. 1999). Those histochemical or cytochemical studies have mostly employed the alkaline diaminobenzidine (DAB) reaction to identify the peroxisomes by detecting the peroxidatic activity of catalase, a marker enzyme for peroxisomes (Fahimi 1969).
Although many peroxisomal proteins have been characterized at the molecular level in recent years, there are only a few reports on the distribution of their mRNA transcripts in different organs, mostly using radioactive ISH because of the relatively low expression levels (Bout et al. 1990; Pollard et al. 1995). Only Reimer and Singh (1990) have reported significant catalase (CAT) mRNA expression in liver and brain during mouse fetal development, using 35S-labeled riboprobes. Recently, Schad et al. (1996) introduced a novel protocol for nonradioactive ISH using digoxigenin-labeled riboprobes on paraffin sections for highly sensitive detection of mRNAs encoding peroxisomal proteins. Thus, mRNAs for CAT and urate oxidase, representing high-abundance transcripts, as well as mRNAs for multifunctional protein 1, acyl-CoA oxidase (AOX), and PMP-70, representing low-abundance transcripts, have been localized in adult rat liver and kidney (for a recent review see Fahimi and Baumgart 1999).
Because of increasing interest in mouse development in conjunction with transgenic mouse models of human diseases, we decided to investigate the localization of peroxisomal proteins and their corresponding mRNAs in fetal and newborn mice by IHC and nonradioactive ISH. In this article we present a protocol to detect two such proteins and the corresponding mRNAs on consecutive sections of fetal and newborn mouse tissues. We demonstrate the presence of CAT and AOX mRNAs and proteins from 14.5 days p.c. (E14.5) to newborn mice (P0.5) in liver, intestine, and skin. CAT protein is detected in small cytoplasmic granules, resembling peroxisomes. An abstract with results of this study was presented at the Annual Meeting of the German Society for Cell Biology (Grabenbauer et al. 1999).
Materials and Methods
Animals
Swiss mice were kept on a normal laboratory diet and water ad libitum. They were housed in cages in compliance with the guidelines for the humane care and use of laboratory animals of the Federal Republic of Germany. Female mice were placed with males overnight and were considered to be 0.5 days p.c. (post coitum) when a vaginal plug was present the following morning.
Fixation and Tissue Processing
Pregnant females of gestational stages E14.5, E15.5, E16.5, E17.5, and E18.5 days p.c. and newborns were anesthesized with diethylether. The fetuses delivered by C-section and newborns were perfused through the heart with 4% freshly depolymerized paraformaldehyde in PBS (10 mM phosphate buffer with 137 mM NaCl and 2.7 mM KCl at pH 7.4). After postfixation for 24 hr by immersion in the same fixative, the animals were sectioned sagittally in two halves and embedded in paraffin (Paraplast Plus; Sherwood Medical, St Louis, MO) using an automated vacuum infiltration tissue processor (Tissue-Tek V.I.P. E300; Sakura Finetek, Torrance, CA). Three-μm sections of whole animals were cut on a sliding microtome (Leica SM 2000 R; Leica Instruments, Nussloch, Germany) and mounted on Superfrost Plus slides (Shandon; Frankfurt/M., Germany).
In Situ Hybridization
After deparaffinization and rehydration, the sections were pretreated with 100 mM HCl and digested for 30 min at 37C with 5–20 μg/ml proteinase K (Sigma; Deisenhofen, Germany) in TE buffer (100 mM Tris, 50 mM EDTA, pH 8.0). The exact concentration of proteinase K was determined empirically for each block (Schad et al. 1996). Subsequently, the sections were postfixed for 5 min with freshly prepared 4% paraformaldehyde in PBS (pH 7.4) and acetylated with freshly prepared 0.25% (v/v) acetic acid anhydride in 100 mM triethanolamine at pH 8.0, followed by dehydration in ethanol and air-drying. The sections were prehybridized for 2 hr at 45C or 58C in a mixture consisting of 50% (v/v) formamide, 50 mM Tris-HCl (pH 7.5), 25 mM EDTA, 20 mM NaCl, 250 μg/ml yeast tRNA, and 2.5 × Denhardt's solution (Sigma).
Synthesis of the digoxigenin-labeled riboprobes for CAT and AOX and their hydrolysis to 200-base fragments were as described previously (Schad et al. 1996). The hybridization mixture contained 5 ng/μl riboprobe, 50% (v/v) formamide, 20 mM Tris-HCl (pH 7.5), 1 mM EDTA, 333 mM NaCl, and 10% dextran sulfate in a total volume of 20 μl per section. Each section was covered by a siliconized coverslip (Hybrislips; Sigma), sealed with rubber cement, and hybridized overnight at 45C or 58C. To remove the excess probe, the sections were washed at 53C with 2 × SSC (standard saline citrate buffer: 300 mM NaCl, 30 mM sodium citrate, pH 7.2) for 30 min and 1 × SSC/50% (v/v) formamide for 1 hr, followed by twice 0.5 × SSC at room temperature (RT) for 10 min and 0.2 × SSC for 10 min. The hybridizations at 58C were washed at a higher stringency with 2 × SSC for 30 min at RT, followed by 2 × SSC and 0.1 × SSC for 1 hr at 65C. Before digoxigenin detection, nonspecific binding sites were blocked with 1% (w/v) blocking medium (Boehringer Mannheim; Mannheim, Germany), and 0.5% (w/v) bovine serum albumin in TBS buffer (100 mM Tris, 150 mM NaCl, pH 7.5). The sections were incubated at 4C overnight with alkaline phosphatase-labeled anti-digoxigenin Fab fragments (Boehringer Mannheim) diluted at the manufacturer's recommendation in the blocking buffer. The staining reaction for alkaline phosphatase was performed at 37C in darkness with a TNM buffer (100 mM Tris, 100 mM NaCl, 50 mM MgCl2, pH 9.5) containing 275 μM nitroblue tetrazolium salt (NBT), 400 μM 5-bromo-4-chloro-3-indolyl phosphate (BCIP), and 1 mM levamisole (Sigma) after the addition of 10% (w/v) 70-100-kD polyvinyl alcohol (Sigma) to enhance the reaction (DeBlock and Debrouwer 1993). Finally, the sections were counterstained with hematoxylin or nuclear Fast Red and mounted with glycerol-gelatin.
Immunohistochemistry
For antigen retrieval and improved accessibility of epitopes, deparaffinized and rehydrated sections were subjected to any one of the following procedures: digestion with 0.01% trypsin (Sigma) in PBS for 10 min; digestion with 0.1% trypsin for 5 min; microwaving in 10 mM citrate buffer at pH 6.0 for 15 min at 720 W in a conventional household microwave oven (Panasonic), followed by 20 min cooling without changing buffer; or 0.01% trypsin for 5 min followed by 15 min of microwaving. Endogenous peroxidase was inhibited by 3% H2O2 for 5 min and nonspecific binding sites were blocked by 0.5% blocking reagent (NEN Life Science; Boston, MA) according to the manufacturer's recommendation. The endogenous biotin was blocked with an avidin-biotin blocking kit (Vector Laboratories; Burlingame, CA), and sections were incubated overnight at 4C with monospecific antibodies to CAT and AOX, which were characterized earlier (Beier et al. 1988). Antigen binding sites were detected by a peroxidase-conjugated biotin–streptavidin system (Extravidin; Sigma) and visualized either by the 3-amino-9-ethylcarbazole medium (AEC; Sigma) followed by mounting in glycerol–gelatin or by a novel peroxidase substrate, Nova Red (Vector) and mounting in Eukitt (O. Kindler; Freiburg, Germany) after dehydration. The nuclei were counterstained with hematoxylin. Control sections were incubated either with non-immune serum or by omitting the primary antibodies.
Immunofluorescence
In addition to carbamazoles, the antigen binding sites were also detected by the use of fluorescein isothiocyanate (FITC)-labeled tyramides (NEN Life Science) according to the manufacturer's specifications. After counterstaining of nuclei with 4′-6-diamidino-2-phenylindole (DAPI), the slides were mounted in Mowiol 4–88 (Hoechst; Frankfurt/M., Germany) with 0.5% N-propylgallate to retard photobleaching.
Illustrations
The light microscopic sections were analyzed using a Leica DMLB microscope (Leica; Wetzlar, Germany). Digitized images were obtained with a color photo scanner (Kaiser; Buchen, Germany) and processed with Adobe Photoshop (version 5.5) on an Apple MacIntosh computer. Fluorescence preparations were examined using a Leica TCS MP confocal microscope (Leica Microsystems; Heidelberg, Germany). DAPI counterstain was excited with a Spectra Physics/Tsunami multi photon laser system (Spectra Physics; Mountain View, CA).
Results
In Situ Hybridization
The mRNAs for CAT and AOX were expressed as early as gestational stage E14.5, increasing until birth and with the highest abundance in newborns. They were co-expressed within the same cells, with a higher signal intensity for CAT mRNA.
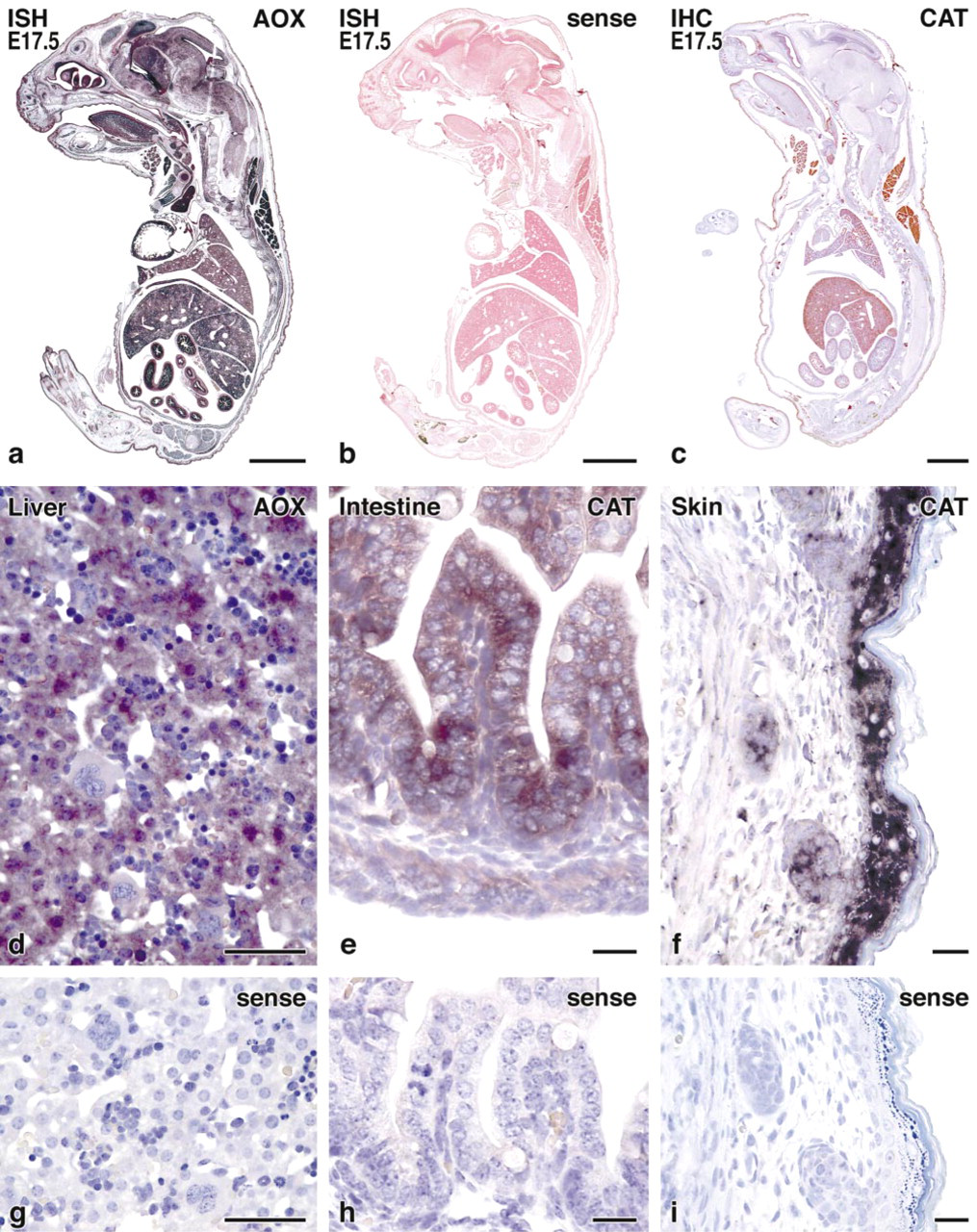
Clear ISH signal enhancement was achieved by the addition of 10% polyvinyl alcohol (70–100 kD) to the alkaline phosphatase BCIP/NBT substrate, allowing detection of mRNAs of peroxisomal proteins also in tissues with low copy numbers (Figure 1a). Control incubations using mRNA (sense) probes for hybridization were consistently negative, confirming the specificity of the method (Figure 1b).
Application of this improved protocol to sagittal sections of complete mouse fetuses revealed the presence of CAT and AOX mRNA in different organs (Figure 1a). Strong signals were obtained in liver, brain, kidney, brown adipose tissue, intestine, and skin. Moderate signals were also detected in thyroid, adrenals, heart, skeletal muscle, respiratory tract, renal tubules, and salivary glands, and weak signals in smooth muscle. Details on cellular and subcellular localization in selected organs (liver, intestine, and skin) are presented below, together with immunocytochemical data.
Immunohistochemistry
IHC analysis of sections incubated with antibodies against CAT showed strong staining with a weaker reaction for AOX in different tissues, corresponding to the localization of mRNAs in parallel sections by ISH (Figure 1c). The use of antigen retrieval by enzyme digestion or microwaving in citrate buffer resulted in strong signal enhancement, with further improvement by the combination of both, weak digestion (5 min with 0.01% trypsin) followed by microwaving (15 min) (Figure 2).
At low magnification, the use of AEC as substrate for the final horseradish peroxidase enzyme reaction showed comparable results to Nova Red (data not shown), but at higher magnifications only the Nova Red substrate showed a punctate staining pattern (Figure 3), contrasting the diffuse cytoplasmic AEC staining (data not shown). The best subcellular resolution was achieved by the use of fluorescein-coupled tyramine as substrate for the final HRP reaction. Further improvements in resolution were obtained by the use of confocal laser scanning microscopy for analysis of sections (Figure 3i). In contrast, the use of directly fluorescent (DTAF-conjugated) secondary antibodies resulted in much lower fluorescence intensity (not shown), emphasizing the strong signal enhancement by the biotin–streptavidin–HRP method combined with catalyzed reporter deposition (CARD) of fluorophorelabeled tyramides. In general, the granular staining pattern for peroxisomes was best obtained with the antibody to CAT, while the AOX antibody gave a rather diffuse cytoplasmic staining (data not shown). No immunoreactivity was found in parallel incubations of control sections when the primary antibody was omitted or replaced by preimmune serum throughout all combinations of secondary antibodies and signal enhancement methods (not shown).
Liver
In the developing mouse liver, the mRNAs for peroxisomal proteins were mainly localized in the cytoplasm of hepatic parenchymal cells and were almost absent in normoblasts and megakaryocytes (Figure 1d). The absence of staining in nuclei and nucleoli and the complete absence of signal in control hybridizations using sense mRNA (Figure 1g) confirmed the specificity of the hybridization. By IHC, peroxisomes are detectable as early as gestational stage E14.5, where the few small hepatocytes were best recognized by the staining of peroxisomes (Figure 3a). In newborn mouse liver, the number and size of hepatocytes increased and they contained distinct granules corresponding to the distribution of peroxisomes (Figures 3b and 3c).
In situ hybridization and immunohistochemistry of sagittal sections of complete mouse fetuses at developmental stage E17.5 (
Intestine
The mRNAs for CAT and AOX in the small intestine were mainly localized in enterocytes and, to a lesser extent, in developing smooth muscle cells (Figure 1e). Regarding the crypt–villous axis, the enterocytes showed similar staining intensity and no significant difference between the villi and crypts. In contrast to the homogeneous cytoplasmic distribution of mRNA, the peroxisomes of enterocytes, as detected by IHC, were mainly localized to the supranuclear area of the cytoplasm (Figure 2c), with an intensification of this phenomenon during development from gestational stage E14.5 to E18.5 (Figures 3d–3d). At higher magnification, we observed a certain degree of heterogeneity in the intensity of CAT immunostaining in different peroxisomes even within the same cells (Figure 3f).
Skin
Figure 1f shows the distribution of CAT mRNA in the skin of a newborn mouse, which is mainly localized in suprabasal layers of epidermis and, to a lesser extent, in hair follicles. The corresponding sense control is negative (Figure 1i). The onset of catalase immunostaining, in contrast to liver and intestine, first appeared weak and diffuse (E14.5–E16.5), and punctate staining of small peroxisomes was detected only at late pregnancy at E18.5 (Figure 3g), with no intensification of staining thereafter (Figures 3h and 3i). Similar to the distribution of mRNA, peroxisomes were also more concentrated in the suprabasal layers of epidermis. Staining of hair follicles for CAT remained diffuse even at birth.
Discussion
The results described here establish for the first time the histochemical and cytochemical localization of peroxisomal proteins and corresponding mRNAs in tissues of fetal and newborn mice at high subcellular resolution. Moreover, in serial sagittal sections of complete animals, we could show the distribution of mRNAs and the corresponding proteins. Whereas the mRNAs were exclusively in the cytoplasm, the proteins were found in fine granules corresponding to peroxisomes. This was made possible by combining our original protocol for ISH (Schad et al. 1996) with an antigen retrieval step for detection of peroxisomal proteins in paraffin sections. In addition, the use of PVA in alkaline phosphatase staining increased the sensitivity of the ISH protocol, and the use of Nova Red substrate improved the detection of peroxisomes by IHC. Several aspects of our protocol should be briefly considered here before the discussion of the role of peroxisomes in fetal and newborn mice.
Fixation and Embedding
The method of perfusion–fixation with aldehydes has been shown to be superior to immersion–fixation, not only for ultrastructural preservation (Fahimi 1967) but also for cytochemistry and IHC (Yokota and Fahimi 1981; Fahimi and Baumgart 1999), and for ISH (Tournier et al. 1987; Schad et al. 1996; Baumgart et al. 2000), and IHC (Yokota and Fahimi 1981; Werner et al. 1996). Here we used 4% freshly prepared paraformaldehyde and omitted glutaraldehyde to avoid stronger masking of epitopes. Perfusion through the heart worked well for newborn and fetal mice, even at E14.5. Care must be taken that the initial rinse with physiological saline to remove the blood cells does not exceed 30 sec, because otherwise mRNA starts to be degraded. The paraffin embedding provided an excellent support for obtaining serial sections of whole animals (Figures 1a–1a) and superior cytological resolution to frozen sections (Tournier et al. 1987; Schad et al. 1996; Baumgart et al. 2000). The use of an automated vacuum infiltration tissue processor resulted in better paraffin penetration, permitting routine serial sectioning below 5 μm.
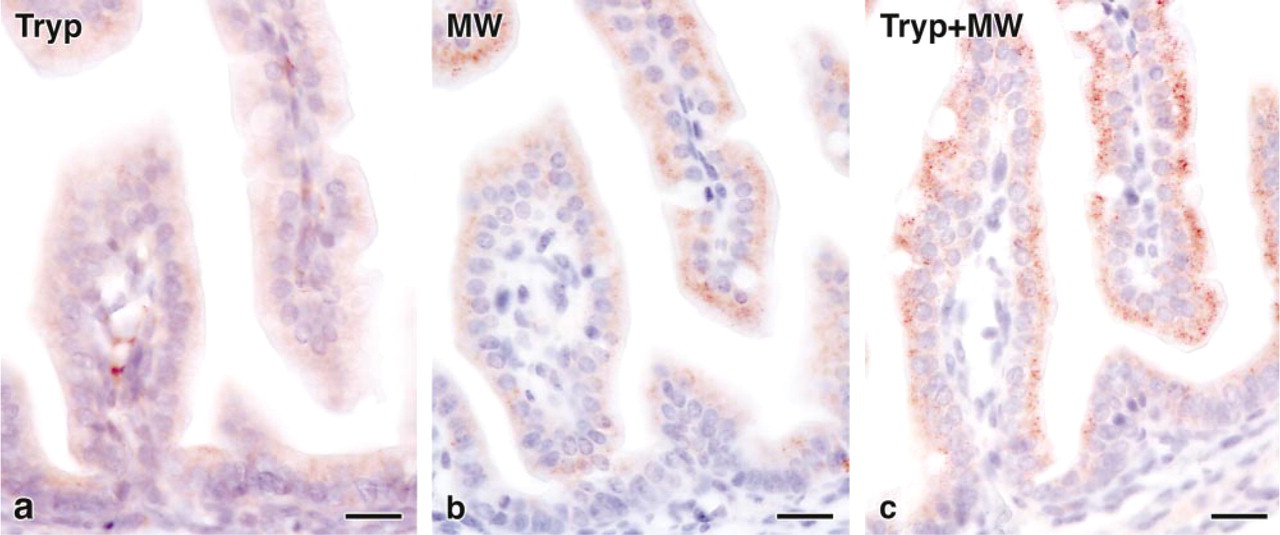
Comparison of different antigen retrieval methods for detection of CAT in the intestine of E18.5 mouse fetus. (
Antigen Retrieval
Paraformaldehyde fixation and paraffin embedding are known to reduce antigenicity caused by crosslinking of epitopes and changes of tertiary and quaternary protein structures (Werner et al. 1996). The most commonly used retrieval techniques in IHC are proteolytic digestion (Huang et al. 1976; Litwin et al. 1988) and high-temperature microwaving (Shi et al. 1991, 1997; Werner et al. 1996). In this study, we found that the combination of both methods, with mild trypsin digestion before microwave heating, resulted in strongly enhanced antigenicity, together with excellent morphology and no background staining (Figure 2).
Improved Detection of ISH and IHC Signals
In enzyme cytochemistry, it is well established that improved localization of enzyme activity can be achieved through the addition of viscosity-increasing polymers to the incubation media, which has also been adapted to visualize ISH signals (Kiyama and Emson 1991; DeBlock and Debrouwer 1993). The addition of PVAs of high molecular weight (70–100 kD) to the NBT/BCIP reaction medium strongly enhanced the alkaline phosphatase reaction and prevented the diffusion of reaction intermediates, thus improving the sensitivity of our method. For detection of low copy number mRNAs, even prolonged substrate incubations (more than 24 hr) were possible without higher background staining.
In IHC, the new HRP substrate Nova Red was superior to the well-established AEC by yielding higher signal intensity and superior optical resolution of enzyme binding sites. Using the new substrate, peroxisomes could be detected by their punctate staining pattern (Figures 2 and 3), whereas AEC as substrate resulted in a more diffuse staining in parallel sections.
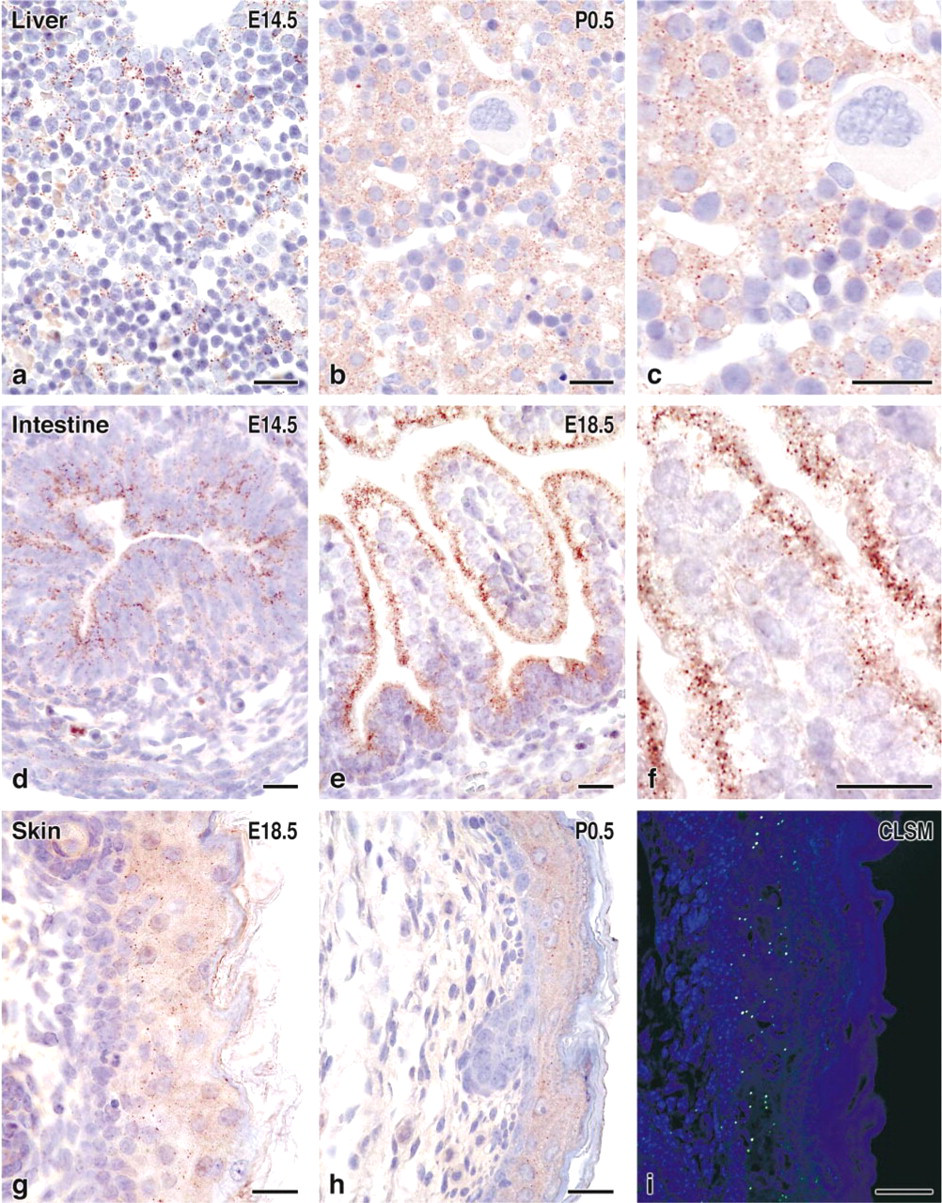
Detection of peroxisomes by CAT immunohistochemistry in liver, small intestine, and skin of fetal and newborn mice. Control incubations with non-immune serum or without primary antibodies were negative (not shown). (
Because the Nova Red reaction product is insoluble in organic media, it was possible to dehydrate the sections and mount them with permanent mounting medium for prolonged storage.
To obtain better microscopic resolution for detection of immunostaining of peroxisomes, we also used fluorescence microscopy. Paraformaldehyde-fixed tissue is known for its high background autofluorescence and, indeed, using fluorescent secondary antibodies we could hardly detect peroxisomes. By using the recently introduced fluorochrome-labeled tyramines as substrates for HRP (van Gijlswijk et al. 1997), however, we obtained very sensitive and precise localization of peroxisomes in skin (Figure 3i). This protocol combines the avidin–HRP approach with the catalyzed reporter deposition (CARD) of fluorescein-labeled tyramides. Furthermore, improved resolution was achieved by the use of a confocal laser scanning microscope (CLSM) (Figure 3i).
Peroxisomes in Fetal and Newborn Mouse Tissues
There have been only a few reports on peroxisomal proteins and their mRNAs in fetal and newborn mouse tissues. Because this report deals only with our findings in three major organs (liver, intestine, and skin), we discuss here our observations in the context of available literature. It is noteworthy that the overall distribution of mRNAs for CAT and AOX corresponds closely to the pattern reported for peroxisome proliferator-activated receptor-α (PPARα) (Braissant and Wahli 1998). This is not surprising because PPARα regulates the transcription of peroxisomal β-oxidation enzymes, particularly that of AOX 1 (Osada et al. 1996).
Liver
Peroxisomes in fetal rat liver were first described by routine electron microscopy (Tsukada et al. 1968). After the introduction of the DAB technique for localization of CAT (Fahimi 1969), peroxisomes were detected as early as E16.5 in mouse (Essner 1969), E14.5 in rat (Stefanini et al. 1985), and at 7 weeks menstrual age in humans (Espeel et al. 1993, 1997). Using radioactive ISH, Reimer and Singh (1990) described the localization of CAT mRNA in fetal mouse liver and brain at E13.5, whereas in whole embryos the earliest biochemical detection was reported as early as with somite formation at Day 8 of gestation (El-Hage and Singh 1990). Our demonstration of peroxisomal proteins and mRNAs in E14.5 mouse fetus concurs with some of the above reports, although we did not attempt to investigate earlier stages of ontogenesis.
Intestine
In the developing intestine of mouse fetus, the number of peroxisomes appears to increase with time, which is consistent with earlier cytochemical and biochemical reports (Pipan and Pšeničnik 1975; Calvert and Menard 1978; Dauça et al. 1996). In contrast to some electron microscopic studies (Calvert and Menard 1978; Dauça et al. 1996) in which elongated peroxisomes in enterocytes were noted, in the present study, despite the limitations of light microscopy, most peroxisomes appeared to be more or less spherical (Figure 3f). The heterogeneity of CAT content in different peroxisomes, even within single cells, that was reported for enterocytes of adult human small intestine (Roels et al. 1991) was also visible even at gestational stage E14.5 (Figure 3d), the earliest stage examined in our study. Regarding the crypt–villous axis in enterocytes of fetal and newborn mice, peroxisomes show a comparable staining intensity from the tip of the villi to the bottom of the crypts without any gradient (Figures 3e–3e), in contrast to peroxisomes of adult rat intestine (Cablé et al. 1993), which show stronger CAT activity in crypts. The detection of peroxisomes and mRNAs encoding peroxisomal proteins in smooth muscle cells of small intestine (Figures 1e and 3d–3e) has not been reported previously, although they have been described in uterus and aorta (Hruban et al. 1972). PPARs in the fetal digestive tract have also been localized mainly to enterocytes (Huin et al. 2000).
Skin
This is, to the best of our knowledge, the first report of epidermal localization of peroxisomal proteins by IHC and corresponding mRNAs by ISH. In agreement with biochemical data of CAT activity (Shindo et al. 1994), we could demonstrate peroxisomes to be localized predominantly in epidermis (Figures 3g–3g), where peroxisomal lipid metabolism could be involved in the development of the fetal epidermal lipid permeability barrier, which is regulated by PPARα and other activators of nuclear hormone receptors (Hanley et al. 1997; Komuves et al. 1998). Further studies on skin peroxisomes should reveal their possible alterations induced by ultraviolet light exposure, where antioxidant enzymes are increased (Shindo et al. 1993), as well as in other conditions associated with altered CAT levels in the skin, e.g., vitiligo (Schallreuter et al. 1991) or progeria (Yan et al. 1999).
In summary, the spatiotemporal distribution of peroxisomal proteins and corresponding mRNAs has been described during the fetal development of mouse liver, intestine, and skin. This was made possible by developing advanced protocols for nonradioactive ISH and IHC on paraformaldehyde-fixed and paraffin-embedded tissues, using improved enzyme substrates, antigen retrieval, and CARD to enhance detection sensitivity and subcellular microscopic resolution. Further studies using these techniques should be helpful in elucidation of cell-specific distribution of peroxisomal—and also non-peroxisomal—proteins and corresponding mRNAs during mouse development, particularly with regard to the wide field of transgenic and knockout mouse models.
Footnotes
Acknowledgments
Supported by The Deutsche Forschungsgemeinschaft (grants Ba 1155/1–3).
We thank Drs U. Deschl and J. Pill, and G. Dietmann (Boehringer Mannheim; now Roche, Mannheim) for logistic support and for supplying pregnant mice, and Prof A. Völkl and Dr A. Schad for many helpful suggestions and discussions. The expert technical assistance of Heike Steininger and Richard Morlang is gratefully acknowledged.