
Abstract
Tyrosine phosphorylation is an important post-translational modification of proteins, essential in many aspects of the cell economy, particularly in signal transduction pathways. Despite the importance of protein tyrosine phosphorylation, the approaches available for molecular cloning remain limited. We have developed a COS cell-based eukaryotic expression cloning procedure for phosphotyrosine-containing proteins by immunocytochemistry of cell monolayers. The approach takes advantage of the low basal levels of tyrosine phosphorylated, robust transient expression, availability of specific antibodies against tyrosine-phosphorylated residues, and rescue of episomal DNA after immunocytochemistry. The technique is validated by cloning the rat proto-oncogene c-fgr in its tyrosine-phosphorylated form out of a rat kidney cDNA library containing over 106 primary recombinants. This technique set the grounds for expression cloning of tyrosine-phosphorylated proteins in eukaryotic cells, and it is anticipated that further modifications and refinements will allow the identification of protein tyrosine phosphatase substrates. (J Histochem Cytochem 48:1097–1101, 2000)
Keywords
Protein tyrosine phosphorylation has been implicated in a myriad of physiological and pathological processes, including embryogenesis, cell differentiation, and cell transformation (for review see Hunter 1998). Given that tyrosine-phosphorylated proteins are very heterogeneous, ranging from structural proteins through cell surface receptor and nuclear transcription factors, it is difficult to devise a specific cloning strategy based on amino acid homology. The major approaches that have been pursued to clone cDNAs encoding phosphotyrosine-containing proteins involve affinity-purification using anti-phosphotyrosine antibodies (Glenney and Zokas 1989) and screening of λGT-11 libraries (Letwin et al. 1988). A more recent cloning strategy using a prokaryote expression and anti-phosphotyrosine antibody as probe has been described (Beeler et al. 1994). We have developed a COS cell-based eukaryotic expression cloning procedure to clone phosphotyrosine-containing proteins. The approach takes advantage of (a) the low basal levels of tyrosine-phosphorylated proteins in many cells, including COS cells, (b) the robust transient expression of transfected cDNAs, (c) the availability of specific antibodies against tyrosine-phosphorylated residues, and (d) the rescue of episomal DNA after immunocytochemistry. The technique is validated by cloning the rat proto-oncogene c-fgr in its tyrosine-phosphorylated form out of a rat kidney cDNA library containing over 106 primary recombinants.
Materials and Methods
cDNA Library Construction
An oligo-dT-primed cDNA library was constructed from kidneys of Sprague-Dawley rats' poly-A+ RNA and was ligated into the BstX I cloning sites of the pcDNAI vector (Invitrogen; Carlsbad, CA). The library was size-fractionated for inserts larger than 1.5 kb and consisted of 3 × 106 primary cDNA recombinants (Invitrogen).
Tissue Culture Techniques
COS MT3 cells that inducibly express the large T-antigen from the metallothionein promoter (Gerard and Gluzman 1985) were obtained from the late Dr. Gluzman (Lederle Labs; Pearl River, NY). The cells were maintained in DMEM (Sigma; St Louis, MO) supplemented with 10% Serum Plus (JRH Biosciences; Lenexa, KS), 100 U/ml penicillin, and 100 μg/ml streptomycin (Sigma).
Transient Transfection
COS MT3 cells at 50–80% confluence were transfected using DEAE-dextran as described previously (Luthman and Magnusson 1983), with some modifications (Golub et al. 1989; Reston et al. 1991). Ten Petri dishes of 15-cm diameter containing 50–80% confluent COS cells (Fisher Scientific; Pittsburg, PA) were washed with PBS, pH 7.4, and treated with 0.05% trypsin and 0.02% ethylenediamine tetraacetic acid (EDTA). Two μl of trypsin was added per dish and incubated for 2 min. Excess trypsin was aspirated and allowed to incubate at 37C until cell detachment occurred (usually 5 min). The cells were resuspended in 4 ml of 400 μg/ml DEAE-dextran (500,000 MW), 0.1 mM chloroquine in DMEM supplemented with 10% Serum Plus, and 4 μg of a rat kidney cDNA library and were incubated at 37C for 2 hr (the time necessary to allow the cells to adhere to the dish). The transfection solution was then aspirated and washed in PBS twice before shocking with 10% dimethyl sulfoxide (DMSO) in PBS for 2 min at room temperature (RT). After two additional washes in PBS, the cells were incubated with medium containing 5 nM phorbol 12-myristate 13-acetate for 30 min at 37C. The medium was aspirated, washed in PBS, and incubated at 37C with complete medium containing 100 μM ZnSO4 until screening (48–72 hr).
COS Cell Screening by Immunocytochemistry
The transfected Petri dishes were treated for 15 min with 1 mM Na3VO4 at 37C, followed by two washes in 5 ml of PBS, and were fixed in absolute ethanol (–20C) for 10 min. After rehydration in PBS (5 min each), the cells were incubated with the anti-phosphotyrosine antibody 4G10 (Upstate Biotechnology; Lake Placid, NY) at a dilution of 1:5000. Cells were then washed three times in PBS (5 min) and incubated with a donkey anti-mouse antibody coupled to HRP at 1:200 dilution for 30 min. After an additional three washes in PBS, a colorimetric reaction was obtained with 4-chloro-1-naphthol and N, N-dimethyl-p-phenylenediamine monohydrochloride for 10–15 min as described (Conyers and Kidwell 1991).
Visualization of Positive Cells and Plasmid Rescue
The dishes were searched at a low magnification (× 40) for positive cells (dark blue) under a dissecting microscope (Leica Microsystems; Deerfield, IL). Nineteen COS cells were judged positive and subjected to plasmid rescue as follows. Dishes were allowed to dry for 20 min at RT and the putative positive cells were encircled (~0.5 cm diameter) by marking the bottom of the Petri dish with a diamond knife, and were processed to the Hirt preparation (Hirt 1967). The circles were filled with 2 μl of a solution of 0.5% SDS, 10 mM EDTA (pH 8.0), and cell solubilization was monitored (about 1 min). The lysate was pipetted into a 1.5-ml micro-centrifuge tube and the remaining cell debris collected by repeating the procedure twice. A final concentration of 0.5 M NaCl was added and the DNA was ethanol-precipitated with 20 μg of glycogen (Roche; Indianapolis, IN) as carrier. The pellet was washed twice with 70% ethanol (500 μl) and resuspended in 5 μl distilled water. Two μl was electroporated into the MC1061/p3 E. coli strain (Invitrogen) [3 × 109 colony forming units (cfu) per μg of plasmid DNA] according to the manufacturer's instructions for the Gene Pulsar Apparatus (Bio-Rad; Hercules, CA) and the transformation was plated in 10 LB/tetracycline 10-cm-diameter plates. Replica plates were obtained in nitrocellulose discs (NEN Life Science; Billerica, MA) cut into eight equal-sized slices, individually incubated with 25 ml LB/tetracycline (10 μg/ml) medium, and allowed to grow overnight. Plasmid DNA was obtained by alkaline lysis (Sambrook et al. 1989). One tenth of the DNA preparation was used in the second round of transfection. The slices were made smaller or, if few colonies per slice, picked individually, and the procedure was repeated until a single clone inducing an elevated level of tyrosine phosphorylation was obtained. The steps involved are illustrated in Figure 1.
Characterization of cDNA
Double-stranded DNA was prepared using a commercially available kit (Qiagen; Santa Clara, CA) and was sequenced by the chain termination reaction according to the manufacturer's instructions for the Sequenase version 1.0 kit (United States Biochemicals; Cleveland, OH). In addition, the cDNA was restriction-mapped with the enzymes Aha 2, HinP 1, Mse I, Pst I, and Stu I (New England Biolabs; Beverly, MA) for further characterization.
Immunoblotting
COS cells transfected with the cloned cDNA (6-cm diameter; 6-well Petri dishes) were solubilized in 500 μl of 1 × Laemmli buffer (62.5 mM Tris-HCl, 2% SDS, 1% β-mercaptoethanol, 10% glycerol, and 0.0025% bromophenol blue) boiled for 5 min, and an aliquot of 10 μl was subjected to 10% SDS-PAGE using a Bio-Rad Miniprotean II apparatus. Proteins were transferred to polyvinylidene fluoride membranes in a semidry transfer apparatus (Millipore; Bedford, MA). Nonspecific binding was blocked with 3% bovine serum albumin in 50 mM Tris-HCl, 100 mM NaCl, and 0.05% Tween-20, pH 8.0. The blots were then incubated with either the anti-phosphotyrosine antibody or an anti-c-fgr antibody (1:2000) (Cambridge Research Biochemicals; Wilmington, DE). After washing three times (5 min per wash) with the same buffer, the membranes were incubated with either donkey anti-mouse antibody conjugated with HRP at 1:5000 dilution for phosphotyrosine detection or with donkey anti-sheep-HRP (1:4000) for c-fgr. Unbound antibodies were removed by washing as above and detection was obtained by enhanced chemiluminescence with an ECL kit (Amersham Pharmacia Biotech; Piscataway, NJ) and exposure to X-ray film.
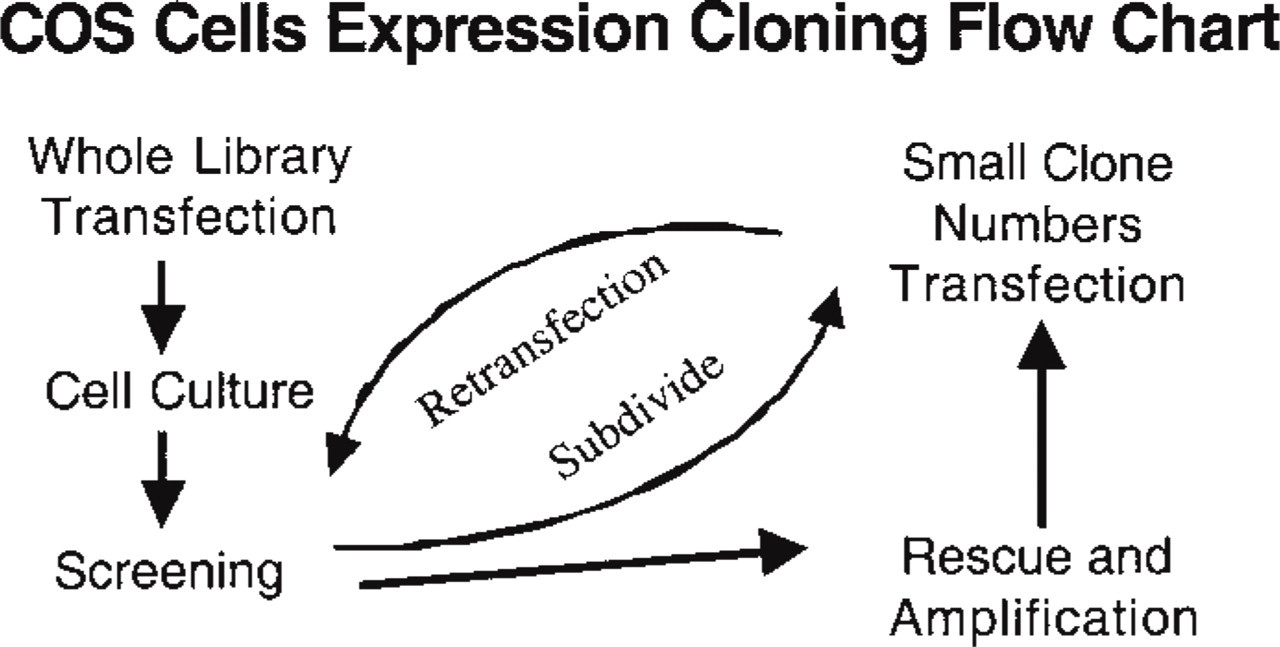
Schematic illustrating the strategies used in the expression cloning. A rat kidney library was constructed in the pcDNA I vector and transfected into COS MT3 cells. COS MT3 cells have been stably transfected with the large T-antigen driven by a metallothionein promoter allowing inducible expression (by adding ZnSO4 to the medium) of the large T-antigen and consequently increasing plasmid replication. After 2–3 days the COS cells were screened for high levels of tyrosine phosphorylation using anti-phosphotyrosine antibodies as described in Materials and Methods. Positive cells, as detected by inspection of the dishes at low magnification after immunocytochemistry, were circled with a diamond knife, and subjected to Hirt preparation as described in Materials and Methods. One third of the Hirt preparation was electroporated in MC1061/p3 cells at the settings recommended by the manufacturers. The electroporated cells were plated in 10 μg/ml of tetracycline-agar plates and grown overnight until the transformed colonies were well visible (usually 16 hr). Nitrocellulose discs were applied to the plates as for plaque hybridization techniques. The discs containing the colonies were divided into eight fractions and each one was incubated with 25 ml of LB/tetracycline medium for 16 hr at 37C. Attention was paid to making pools that do not exceed 500 colonies. In our experience, pools smaller than 500 colonies are easy to identify because several COS cells are usually positive per 10-cm-diameter dish. The master plates were incubated for an additional 6 hr and kept at 4C until the results of the secondaries were obtained. Pools from the plate that scored positive cells were subdivided into smaller groups, usually 10 or, if that fraction contained few colonies, plasmid preparations were made from individual colonies. The procedure was repeated until a single clone was obtained.
Immunoprecipitation
COS cells were lysed in 1 ml immunoprecipitation buffer (20 mM Tris-HCl, 150 mM NaCl, 0.5% Nonidet P-40, 0.05% Tween-20, 10 μM phenylmethylsulfonyl fluoride, and 10 mM NaF, pH 8.0) for 10 min on ice. The lysate was vortexed three times (10 sec each) and was cleared by a brief centrifugation. After incubation with 50 μl of agarose-coupled anti-phosphotyrosine antibody for 16 hr at 4C, the agarose beads were sedimented by brief centrifugation and washed by resuspending and pelleting three times with 1 ml of immunoprecipitation buffer. One hundred μl of 2 × Laemmli buffer was added to the agarose pellet and boiled for 5 min. Ten μl was fractionated on a 10% SDS-PAGE and processed for immunoblotting using the anti c-fgr antibody as described above.
Results
The modifications introduced into the transfection method, i.e., the use of Serum Plus-supplemented medium instead of regular bovine serum, trypsinization (Golub et al. 1989), and a short treatment with phorbol ester (Reston et al. 1991), improve transfection efficiency (data not shown). Pilot experiments indicated that, under the conditions set for immunocytochemistry, no positive cells were detectable in mock-transfected cells. It was also confirmed that fixation in methanol or ethanol allowed plasmid rescue from transfected COS cells (Seed 1995). In the first screening of 10 Petri dishes, 19 cells were judged positive and one clone (Figure 2a) was obtained after three cycles of re-transfection (Figure 2b). The remaining positive cells in the primaries did not rescue positive cells in the secondaries and they were not retried at this time.
Sequence Analysis of the Cloned cDNA
The cDNA clone had an insert of 1.9 kb. Its nucleotide sequences from over 350 bp at either the 5′ and 3′ flanking regions reveal that the cDNA encodes the rat c-fgr tyrosine kinase (accession number X57018.1). Restriction mapping confirmed the presence of restriction sites and fragment sizes for Aha 2, Pst I, Mse I, Hinp I, and Stu I, as predicted from the restriction mapping of the rat c-fgr sequence (accession number X57018.1).
Immunoblotting and Immunoprecipitation
Lysates of COS cells untransfected and transfected with the cloned cDNA were subjected to immunoblotting. In transfected cells only one major tyrosine-phosphorylated band of 55 kD was detected, confirming that the cloned cDNA increases tyrosine phosphorylation in COS cells (Figure 3) and that the 55-kD protein corresponds to a band of the same molecular weight detected with the anti-c-fgr antibody (Figure 3), indicating that the tyrosine kinase is autophosphorylated.
Immunoprecipitation with phosphotyrosine antibodies and blotting confirmed that the major tyrosinephosphorylated protein is the 55-kD c-fgr (Figure 3).
Discussion
The COS cell system is probably the most popular transient eukaryote expression system allowing rapid characterization of new clones. As a cloning approach, it was first used to clone GM-CSF by a functional assay in polls of transfected COS cells (Wong et al. 1985). Later, new strategies were developed to meet the needs and/or skills of each investigator, including panning (Seed and Aruffo 1987), ligand radioautography (Munro and Maniatis 1989; Schaeffer et al. 1991), fluorescent-activated cell sorting (Dell'Arciprete et al. 1996), and immunocytochemistry (Horst et al. 1991; Metzelaar et al. 1991; for review see Seed 1995). In this study we demonstrate the direct isolation of a 1.9-kb cDNA clone encoding a phosphotyrosine-containing protein corresponding to the translated region of the rat tyrosine kinase c-fgr. The proto-oncogene c-fgr is a member of the src family of cytoplasmic tyrosine kinases exclusively expressed in the hematopoietic system (Sharp et al. 1989; Yi and Willman 1989; Sartor et al. 1992). Its expression in different organs has not been addressed in this study but it is likely that the c-fgr clone was originated from peripheral blood or renal resident macrophages at the time of Poly-A+ RNA preparation. The cloning was accomplished by transfecting cDNAs from a whole rat kidney library cloned into an expression cloning vector and rescuing COS cells containing a high level (detectable) of tyrosine phosphorylation after immunocytochemistry of cell monolayers. We have used an anti-phosphotyrosine antibody to detect COS cells expressing high levels of tyrosine phosphorylation because substrates for tyrosine kinases are of a large range of proteins from cytoplasmic and nuclear localization to cell surface growth factor receptors and structural proteins. Because there is no suitable consensus sequence to design oligonucleotides for PCR-based cloning strategies except within families of proteins, the expression cloning approach using anti-phosphotyrosine antibodies becomes an attractive one. There is, however, a screening to determine the consensus sequences for tyrosine kinases based on degenerated peptide libraries (Songyang and Cantley 1998).
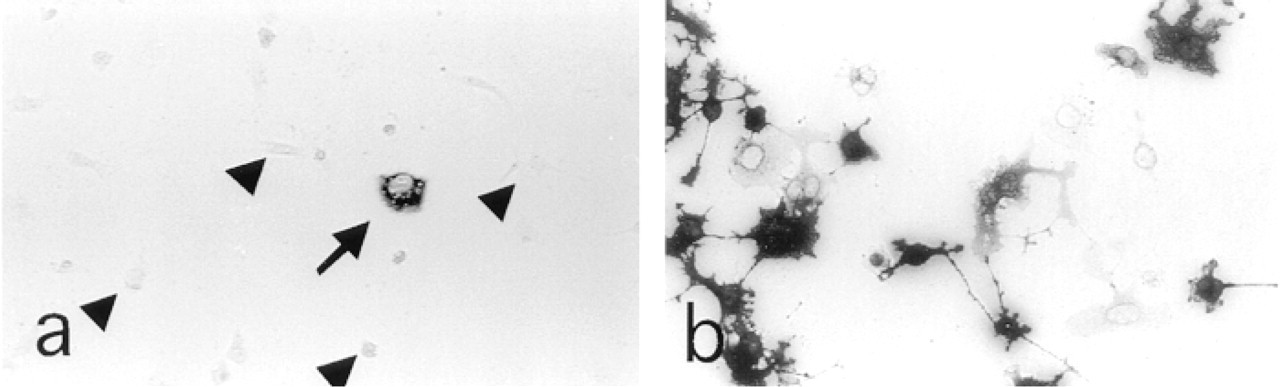
Immunocytochemistry with an antiphophotyrosine antibody of COS cells transfected with a rat cDNA library. (
Compared to the approaches described before for affinity purification (Glenney and Zokas 1989) and λGT11 (Letwin et al. 1988), the eukaryote-based expression cloning described here has the advantage of allowing combination of transient and stable transfections as, for example, combinations of substrates and/or kinases. In this regard, Aaronson's group (Beeler et al. 1994) describes a prokaryote system in which a novel PTP was cloned by expressing in bacteria the kinase domain of the keratinocyte growth factor receptor and infecting the bacteria containing the phagemide together with a fibroblast cDNA library and identifying plaques with decreased phosphotyrosine immunoreactivity. Expression of a combination of protein tyrosine kinases and protein tyrosine phosphatases (PTPs) has also been used to address PTP enzymatic specificity (Lammers et al. 1993). One such example would be to clone specific substrates for PTPs by expressing a library into COS cells, or any other suitable host, together with substrate-trapping mutants of the PTP of interest (Flint et al. 1997). In this case, cells expressing high levels of the substrate will remain in the tyrosine-phosphorylation form because the PTP mutant will associate to its substrates without tyrosine dephosphorylation (Sun et al. 1993; Flint et al. 1997). Consequently, COS cells that accumulate the putative substrate in the tyrosine phosphorylated form may be detected with anti-phosphotyrosine antibody staining.
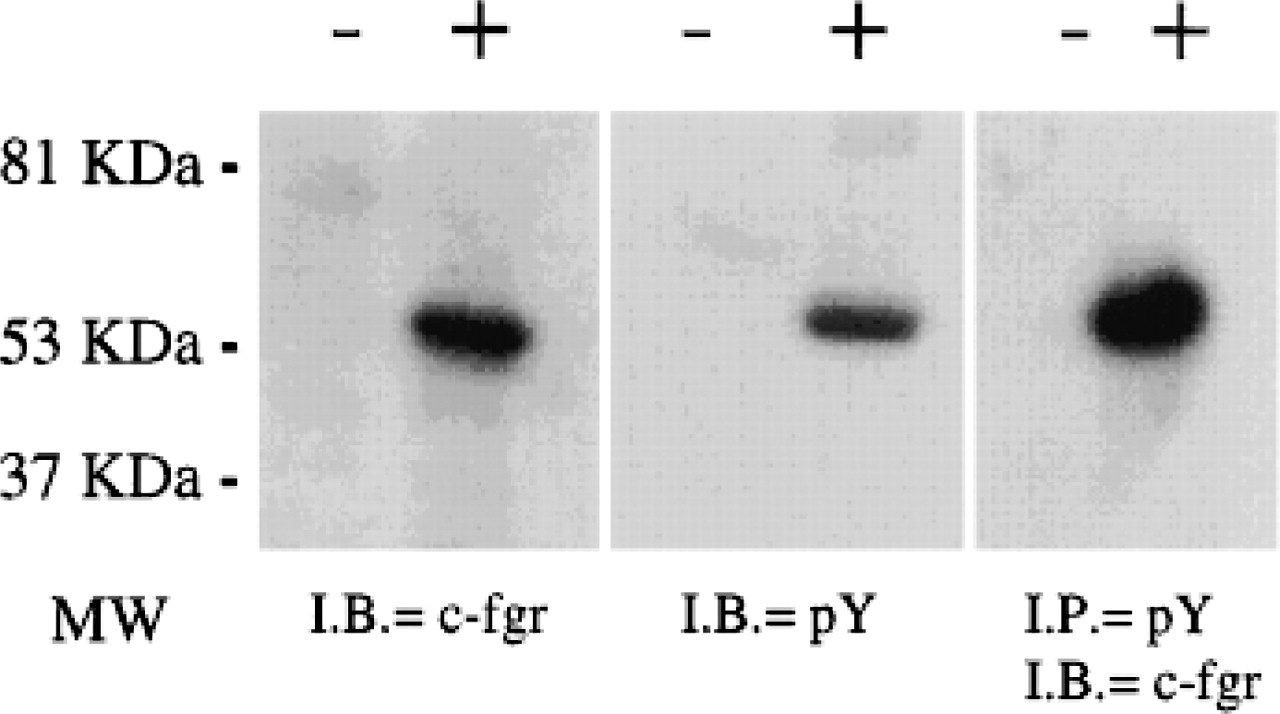
Immunoblotting and immunoprecipitation of COS cell lysate. Lysates from untransfected (–) and transfected (+) COS cells with the c-fgr cDNA clone were immunoblotted (I.B.) with anti c-fgr antibodies (c-fgr) (left), anti-phosphotyrosine (pY) (middle), or lysates were immunoprecipitated (I.P.) (right) with an anti-pY antibody and immunoblotted with anti c-fgr antibody. MW, molecular weight.
The technique described here, despite its flexibility, has the disadvantage of not having been standardized as have prokaryote expression cloning and yeast interaction trapping cloning, for both of which kits are available. However, the COS cell expression cloning has proved useful in situations where more classical cloning techniques may have failed, e.g., the Type 1 angiotensin II receptor (Murphy et al. 1991; Sasaki et al. 1991), angiopoietin-1, a ligand for the TIE-2 receptor (Davis et al. 1996), and the actual slow progress in the identification of in vivo PTP substrates.
Footnotes
Acknowledgments
We wish to thank Drs Charles Homcy and Thomas Quertermous for encouragement and support.