
Abstract
Immunoreactive cystic fibrosis transport regulator (CFTR) proteins in human sweat ducts has been documented but CFTR expression in the secretory coil has remained uncertain. Using monoclonal antibodies (MAbs) against epitopes in the R-domain and C-terminus, we observed the following: Formalin fixation masks the CFTR epitopes but the epitopes are exposed by treatment with urea and heat (antigen retrieval). Pen-Fix fixation preserves CFTR epitopes. The secretory coil also expresses CFTR epitopes for the R-domain and C-terminus. An MAb against C-terminus amino acids 1466-1480 coupled to keyhole limpet hemocyanin (MAb WC) stained dark cells predominantly. Staining by MAbs against the C-terminus was completely blocked by a C-terminus peptide. mRNA for CFTR was amplified by RT-PCR in both the duct and the secretory coil. In situ hybridization for CFTR mRNA after 3SR amplification indicates that mRNA is localized in the dark cells and perhaps also in the clear cell cytoplasm near the secretory coil. mRNA is present in both the luminal and basal duct cells. We conclude that CFTR is expressed equally well in both the duct and the secretory coil, suggesting that cAMP-dependent Cl- channels are involved in regulation of sweat secretion and duct absorption.
T
In 1989, the CF gene was identified (Riordan et al. 1989; Rommens et al. 1989) and in rapid succession its encoded protein (CFTR) has been identified as a cAMP-dependent Cl- channel protein (Drumm et al. 1991; Collins 1992; Welsh and Smith 1993). Identification of immunoreactive CFTR protein has offered promise in understanding the pathogenesis of CF because immunoreactive CFTR was localized in the apical domain of the luminal cells in non-CF but not in CF sweat ducts (Kartner et al. 1992).
Whereas CFTR staining of the sweat duct was consistently observed (Cohn et al. 1991; Crawford et al. 1991; Kartner et al. 1992), that of the sweat secretory coil was not. Kartner et al. (1992) found little evidence for CFTR staining in the secretory coil. Cohn et al. (1991) observed that CFTR staining in the secretory coil was less prominent than in the duct. Crawford et al. (1991) noted dense duct staining but made no mention of staining in the secretory coil.
We sought to establish whether CFTR is absent or is less prominently expressed in the human eccrine secretory coil than in the duct, as these reports suggested. In this study we reexamined the CFTR expression in human secretory coils using two monoclonal antibodies (MAbs) against the C-terminus (Genzyme, Cambridge, MA; and a gift from Dr. Michael Welsh, University of Iowa) and an MAb against the epitope in the R-domain (Genzyme) (Gregory et al. 1990; Denning et al. 1992). In addition, RT-PCR for CFTR transcription was compared between freshly dissected ducts and secretory coils. In situ hybridization after the self-sustained replication-based amplification (3SR) (Zehbe et al. 1994) was also performed to localize CFTR mRNA in the sweat gland.
Materials and Methods
Sample Preparations
Recruitment of human subjects and procurement of surplus skin samples from cutaneous surgery followed the institutional human subject guidelines (University of Iowa, Iowa City, and Human Gene Therapy and Cancer Research Institute, Des Moines, IA). For mRNA isolation, skin specimens were biopsied from four male volunteers, ages 18-35 years, under local block anesthesia with 1% lidocaine. The excised tissues were blotted to remove blood, sliced into thin sections, and immediately washed in several changes of cold (approximately 10C) modified Krebs-Ringer bicarbonate solution (KRB) containing (in mM): 125 NaCl, 5 KCl, 1.2 MgSO4, 1.0 CaCl2, 25 NaHCO3, 1.2 NaH2PO4 (prepared by mixing 1/10th volume of each of the 10 × stock solutions), 5.5 mM glucose, and 0.05% bovine serum albumin (BSA), at pH 7.48, and 5% CO2/95% O2. Single sweat glands were dissected out under a stereomicroscope using sharp forceps in a dissection chamber kept at 14C (Sato and Sato 1981, 1984). About 20-25 sweat glands were isolated from each biopsy specimen. The ducts and the secretory coils were further dissected as previously described (Sato and Sato 1984).
Four methods of tissue fixation/preparation were employed: traditional 10% buffered formalin-fixed paraffin sections; formalin-fixed paraffin-embedded sections subsequently treated by urea in microwave heat for antigen retrieval (Shi et al. 1993); Penn-Fix-fixed (Richard-Allan Scientific; Kalamazoo, MI) sections; paraffin-embedded sections (without antigen retrieval); and routine cryosections of liquid nitrogen-frozen skin specimens. All skin samples were fixed or frozen immediately after removal from the skin.
Antibodies for CFTR
Three MAbs were used, one MAb against an epitope in the R-domain and two MAbs directed against the C-terminus. The MAb directed against the R-domain (residues 729-736) was purchased from Genzyme (hereafter called MAb GR). This MAb was made to a β-galactosidase fusion protein (Gregory et al. 1990). The mouse MAb against the C-terminal epitope (Genzyme) was made to a glutathione S-transferase fused to amino acid residues 1477-1480 (MAb GC). The second MAb for the C-terminus epitope [kindly provided by Dr. Michael Welsh, University of Iowa (MAb WC)] was made to residues 1466-1480 fused with keyhole limpet hemocyanin (Gregory et al. 1990).
Preparation of Histological Sections
Paraffin sections were dewaxed, rehydrated, rinsed in Trisbuffered saline (TBS), with or without treatment in 0.3% Triton for 15 min, and washed in phosphate Ringer (PBS) for 5 min. Because conventional formalin-fixed paraffin sections consistently yielded negative or very poor staining for CFTR in our hands, we elected to combine the antigen retrieval method (Shi et al. 1993) with minor modifications on formalin-fixed paraffin sections in an attempt to expose antigenic sites. Dewaxed, rehydrated sections were placed in a 5 M urea solution kept at 60C for 5 min, then boiled in a microwave oven (Toshiba or Sanyo model) for 5 min. Sections were then left at room temperature (RT) for 5 min and washed in distilled water five times before incubation with primary MAbs. Cryosections were air-dried for 15 min, fixed in acetone for 10 min at RT, and treated with 0.23% (w/v) periodic acid for 10 min at RT to suppress endogenous peroxidase. The sections were blocked in TBS-gelatin blocking buffer (0.5% BSA, 5% horse serum, and 1% Sigma Fish Gelatin in TBS, pH 7.4) for 2 hr at RT and incubated in a 1:20 or 1:100 dilution of MAbs (for all MAb GC, GR, and WC) at 4C overnight. After washing, sections were incubated with a biotinylated anti-mouse Ig at 1:200 for 3 hr at RT. Washed sections were then incubated with a 1:20 dilution of ExtrAvidin-Peroxidase Staining kit (Sigma; St Louis, MO) for 2 hr at RT and the staining visualized using Sigma FAST. To examine the specificity of immunohistochemical staining, we used two negative controls: sections without primary MAbs (which all yielded negative staining) and sections incubated with MAbs and a corresponding peptide that blocks the epitope in the C-terminus (BSA-Nle-A-L-K-E-E-T-E-E-E-V-Q-D-T-R-L-OH; kindly provided by Dr. Michael Welsh). Sections were incubated with primary MAbs overnight in a humid chamber with or without the blocking peptide at 0.2 mg/ml (the ratio determined to be optimal by a dose-response study conducted in Dr. Welsh's laboratory; personal communication). Blocking peptides for the R-epitope were not available for this study.
In Situ 3SR
The basic principle and methods for self-sustained sequence replication-based amplification (3SR) have been described by Gingeras et al. (1990) and its application for in situ localization of mRNA by Zehbe et al. (1994). Cryosections were fixed with 4% paraformaldehyde in PBS for 2 min, washed two times with PBS, and permeabilized with saponin-EDTA (0.1% saponin + 1 mM EGTA in PBS) for 30 min at RT. Sections were postfixed with 4% paraformaldehyde in PBS, washed in 0.2% glycine in PBS, and rinsed in PBS. The sections were dehydrated in graded alcohol, air-dried, and processed for 3SR. The 5′ primer for in situ 3SR is (nucleotide position 1389-1409) AAAAACTTCTAATGGTGATGA. The T7 promoter sequence (lower case) is tagged to the 3′ primer, i.e., aatttatacgactcactataggga-CAGTTTACAGACACAGCTTTCA (position 1893-1914). The 3SR reagents contain 1 mM dNTP, 7 mM rNTP, 1 μM each of 3SR primers, 20 mM Tris-HCl, pH 8.0, 30 mM MgCl2, 10 mM dithiothreitol (DTT), 4 mM spermidine, and DEPC-treated double-distilled water. The 3SR enzyme mixture was prepared in a plastic tube on ice by mixing 1.4 μl (30 U) of avian myeloblastosis virus (AMV) reverse transcriptase (RTase), 5 μl (100 U) of T7 RNA polymerase, and 3 μl (3 U) of RNase H (all from Boehringer; Mannheim, Germany) and 10 μl 3SR reagents. The cryosections were incubated with 22.5 μl 3SR reagents at 65C for 1 min, followed by annealing at 42C for 2 min. Amplification by 3SR was then performed by applying 4.8 μl enzyme mix to sections and incubating for 2 hr at 42C in a humidified chamber. The sections were fixed in 2% glutaraldehyde in PBS for 5 min and dehydrated in graded alcohol. Negative control slides were treated with RNase A and T (200 μg/ml).
Preparation of DIG-PCR Probe and Detection of 3SR Amplicons in In Situ Hybridization
The probe for in situ 3SR was prepared with a polymerase chain reaction (PCR) using a set of primers: GTTTTCCTGGATTATGCCTGGCAC (CFTR position 1611-1634) and AGAAGCGTCATCAAAGCATGCCAAC (position 1684-1708). A 20-μl reaction mixture included 50 mM KCl, 10 mM Tris-HCl, pH 8.3, 1.5 mM MgCl, 200 μM each dATP, dCTP, and dGTP, 130 μM dTTP, 130 μM DIG-11-dUTP (Boehringer Mannheim) 1 μM of each primer, 5 μl cDNA template (for CFTR), and distilled water. A total of 35 cycles of PCR (annealing 1 min at 55C, extension 2 min at 72C, and denaturation 1 min at 93C) were carried out. Final extension was performed at 72C for 7 min. The PCR product was precipitated in ethanol and resuspended in Tris-EDTA. Fifteen μl probe mix containing 50% formamide, 10% dextran sulfate, 5 × SSC, E. coli tRNA (1 μg/(μl), 10% bovine serum albumin (BSA), herring sperm DNA (1 μg/μl), and 0.5 μg/μl digoxigenin-labeled PCR probe were applied to the air-dried slides (previously processed for 3SR amplification) and denatured for 10 min at 95C. Hybridization was performed in a preheated humidified chamber at 42C overnight, followed by three posthybridization washes in 2 × SSC at RT for 5 min each and rinsing in TBS-gelatin (Zehbe et al. 1994). Digoxigenin-labeled hybridization sites were detected at 4C overnight by incubation of a mouse MAb against digoxigenin (Zehbe et al. 1994). After three washes in TBS-gelatin, the slides were subjected to anti-mouse IgG 5-nm colloidal gold followed by silver enhancement (SilvEnhance-LM kit; Zymed, San Francisco, CA).
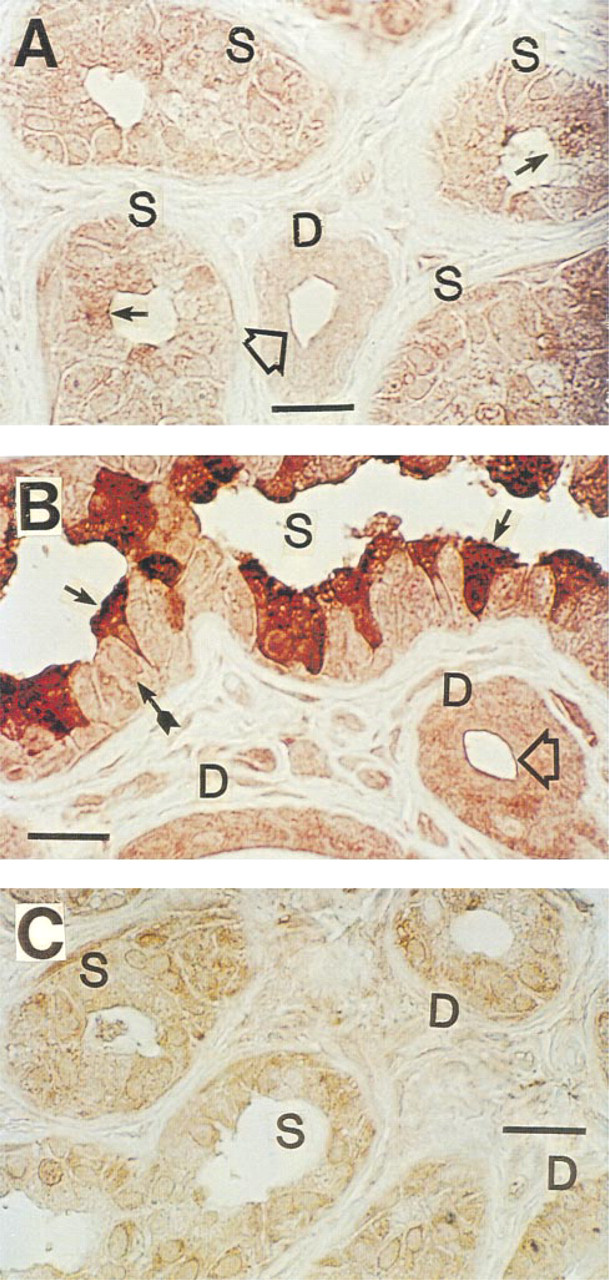
Immunohistochemical staining of CFTR in human eccrine sweat gland after antigen retrieval with urea. (
Isolation of RNA and Detection of CFTR mRNA by RT-PCR
RNA was extracted by incubating in each tube 15 isolated secretory coils, 15 ducts, and eight whole glands at 45C for 1 hr in a digestion mixture containing 6 mg/ml proteinase K, 1 M guanidium thiocyanate, 25 mM 2-mercaptoethanol, 0.5% N-lauroylsarcosine, 20 mM Tris-HCl, pH 7.5 (Stanta and Schneider 1991). After extracting nucleic acids by the phenol/chloroform/isoamyl alcohol (PCI) mixture twice, 0.1 volume 8 M LiCl, 40 μg/100 μl glycogen, and 0.7 volume chilled isopropanol were added to the nucleic acid fraction. The tube was placed on dry ice overnight and centrifuged. The pellets thus obtained were dissolved in 100 μl water and RNA purified by digesting contaminating chromosomal DNA with 40 U/tube RNase-free DNase and RNase inhibitor (Promega; Madison, WI) at 37C for 1 hr. Enzymes were removed by repeating the PCI extraction and the purified RNA reconstituted in water for RT-PCR. mRNA was reverse-transcribed to synthesize the first-strand cDNAs using the oligo-dT or random primers according to instructions for the GeneAmp PCR kit (Perkin-Elmer Cetus; Norwalk, CT). The sequence corresponding to CFTR was amplified by PCR between nucleotides 1389 through 1914 using a set of 25-mers. Amplification was performed on a programmable thermal cycler (Perkin-Elmer Cetus) with 35 cycles of denaturation at 95C for 1 min, annealing at 55C for 1 min, and elongation at 72C for 3 min. Ten-μl aliquots of the reaction mixture were electrophoresed in a 1% agarose gel and visualized by ethidium bromide staining. Because no band was visually identified around the predicted size of the amplified product of 526 bp, a second nested PCR was performed using a set of 25-mers between nucleotides 1611 and 1710, with a predicted amplified fragment of 100 bp. Tubes without RNA (blank) and without RT were simultaneously run as negative controls to detect genomic DNA contamination and other artifacts.
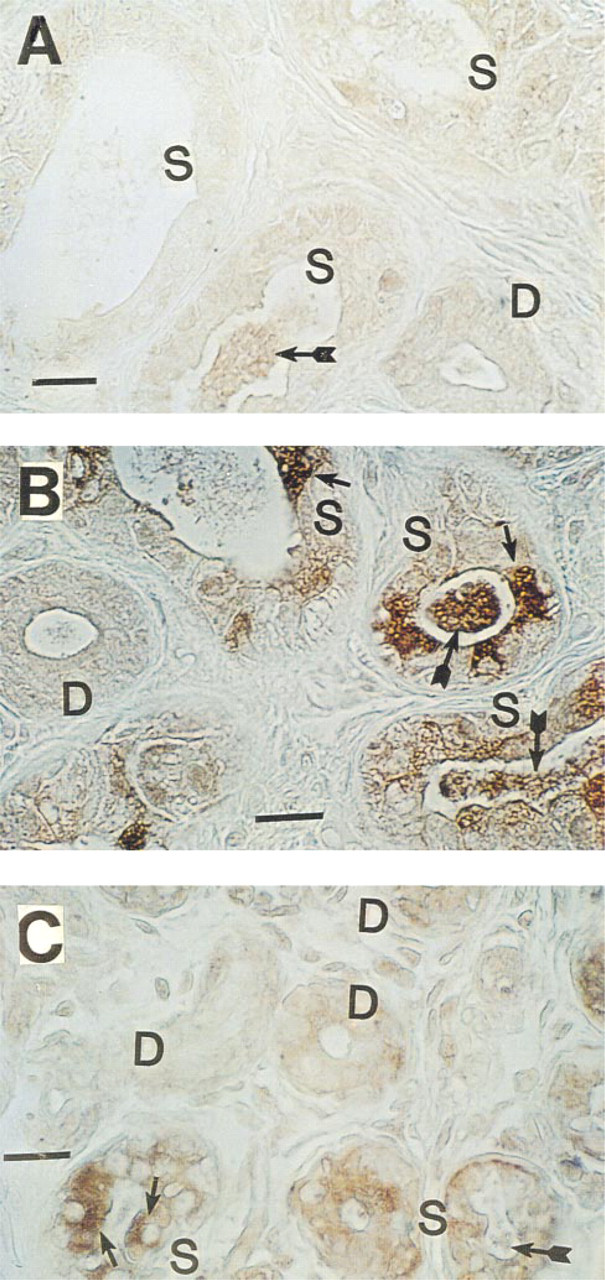
Immunohistochemical staining of CFTR in Pen-Fix-fixed, paraffin-embedded human eccrine sweat gland (without antigen retrieval). (
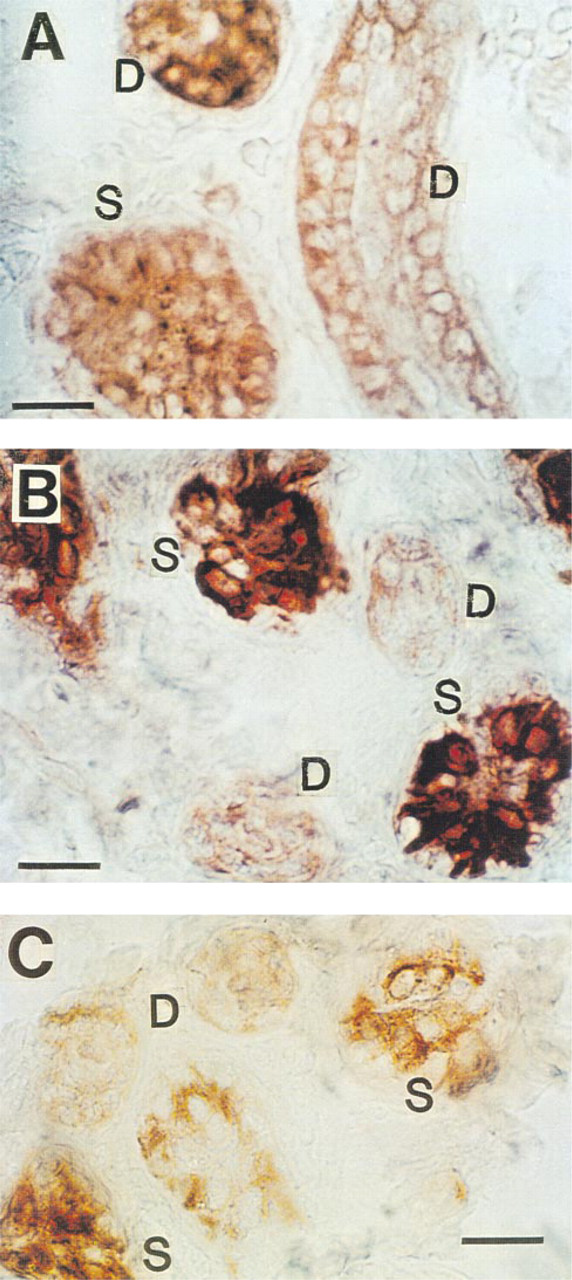
Immunohistochemical staining of CFTR in cryosections of human eccrine sweat gland. (
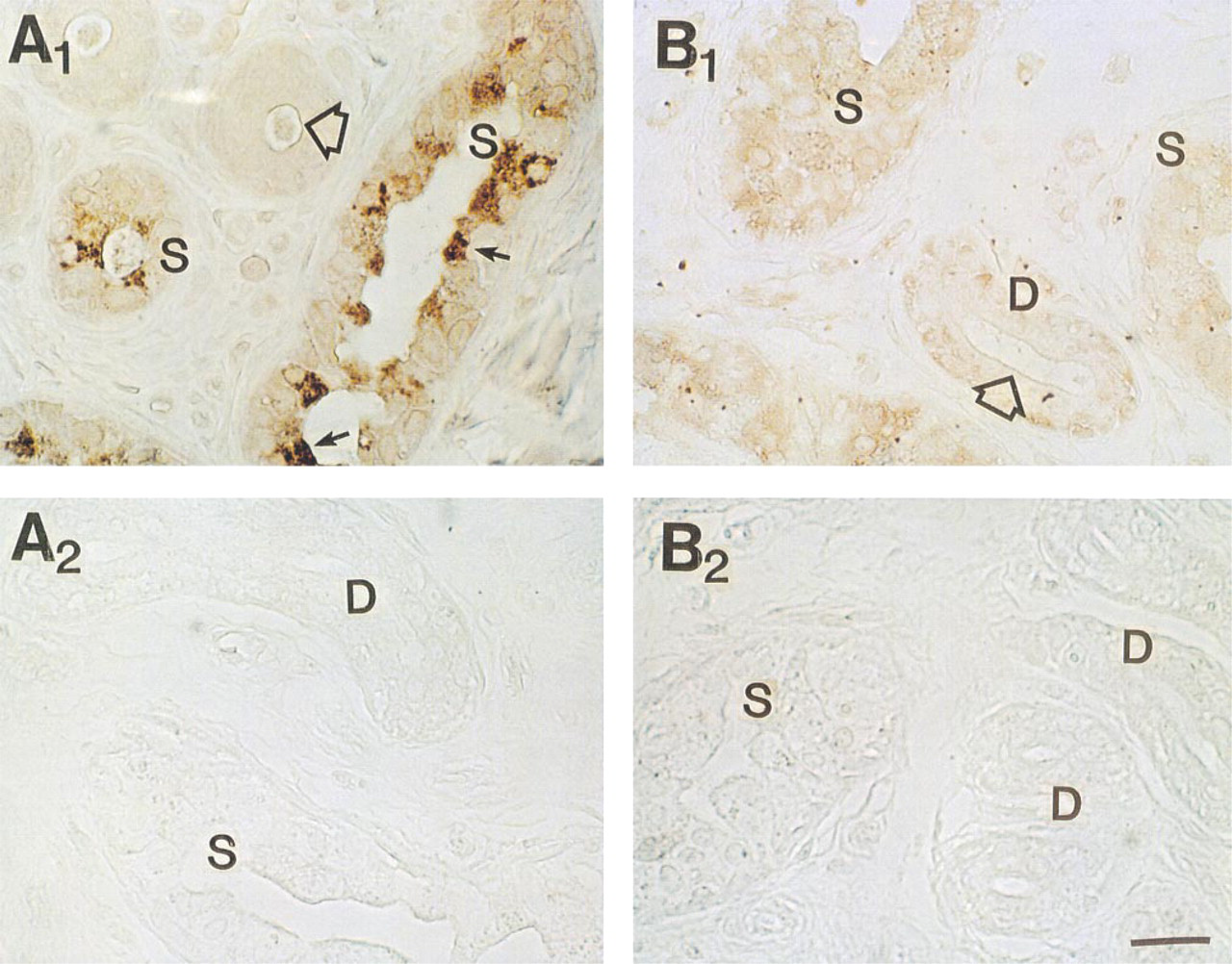
Effect of peptide blocking on MAb staining of CFTR C-terminus in human sweat gland. (
Reagents
All other reagents were obtained from Sigma unless otherwise noted.
Results
In agreement with previous authors (Kartner et al. 1992), conventional formalin-fixed, paraffin-embedded sections stained CFTR very poorly with all MAbs tested (not shown). However, after antigen retrieval with 5 M urea (Shi et al. 1993), consistent staining for CFTR was observed (Figure 1), suggesting that formalin fixation had masked the CFTR epitopes (presumably by forming methylene bridges) for recognition by MAbs employed in this study. Significant differences in the CFTR staining pattern were observed with different MAbs. As shown in Figure 1A, MAb GR stained cytoplasm as well as the cell membrane of all cell types, although some dark cells (small arrows) were stained slightly more densely than other types of cell. In Figure 1B, cytoplasm and membrane of dark cells are most densely stained with MAb WC. Duct cells and clear cells were stained moderately. In Figure 1C (MAb GC), all the cell types (clear, dark, myoepithelial, and duct cells) took up staining to a similar extent, but the dark cell-predominant staining pattern seen in Figure 1B was not consistently observed. This observation was somewhat unexpected because MAb WC (Figure 1B) and MAb GC (Figure 1C) are presumed to recognize the same epitope (i.e., the last four C-terminus amino acid residues, D-T-R-L) in CFTR. In contrast to simple formalin fixation, positive staining could be observed when the tissue was fixed in Pen-Fix (a mixture of formalin and a buffered dehydrant) followed by routine paraffin embedding without antigen retrieval. Figure 2 demonstrates mildly positive staining with MAbs at 1:100 dilution in Pen-Fix-fixed, paraffin-embedded sections in patterns similar to those seen in Figure 1. Of note, the prominent staining of dark cells by MAb WC suggests that the dense dark cell staining seen in Figure 1B is not an artifact of urea treatment. With MAb GC, dark cells were slightly more densely stained than other types of cell. Interestingly, sweat protein or cell debris in the lumen (thin long arrows) were also stained with all the MAbs, and with MAb WC in particular (Figure 2). To further examine the specificity of MAb staining for CFTR, especially in paraffin-embedded sections, cryosections were also used after fixation with acetone (Figure 3). All MAbs stained both the secretory coil cells and duct cells. MAb WC densely stained not only the dark cells but the clear cells as well (Figure 3B). MAb WC also stained the secretory coil more densely than the duct cells (Figure 3B). The specificity of immunohistochemical staining with the MAb for the CFTR C-terminus was examined using a 16-amino-acid peptide that includes the MAb-targeted epitope (C-terminal four amino acids). In both Pen-Fix-fixed, paraffin-embedded sections and cryosections, the peptide completely abolished staining by MAb WC (Figures 4A2 and 5B) and MAb GC (Figure 4B2), indicating that both MAbs reacted with the C-terminus epitope.
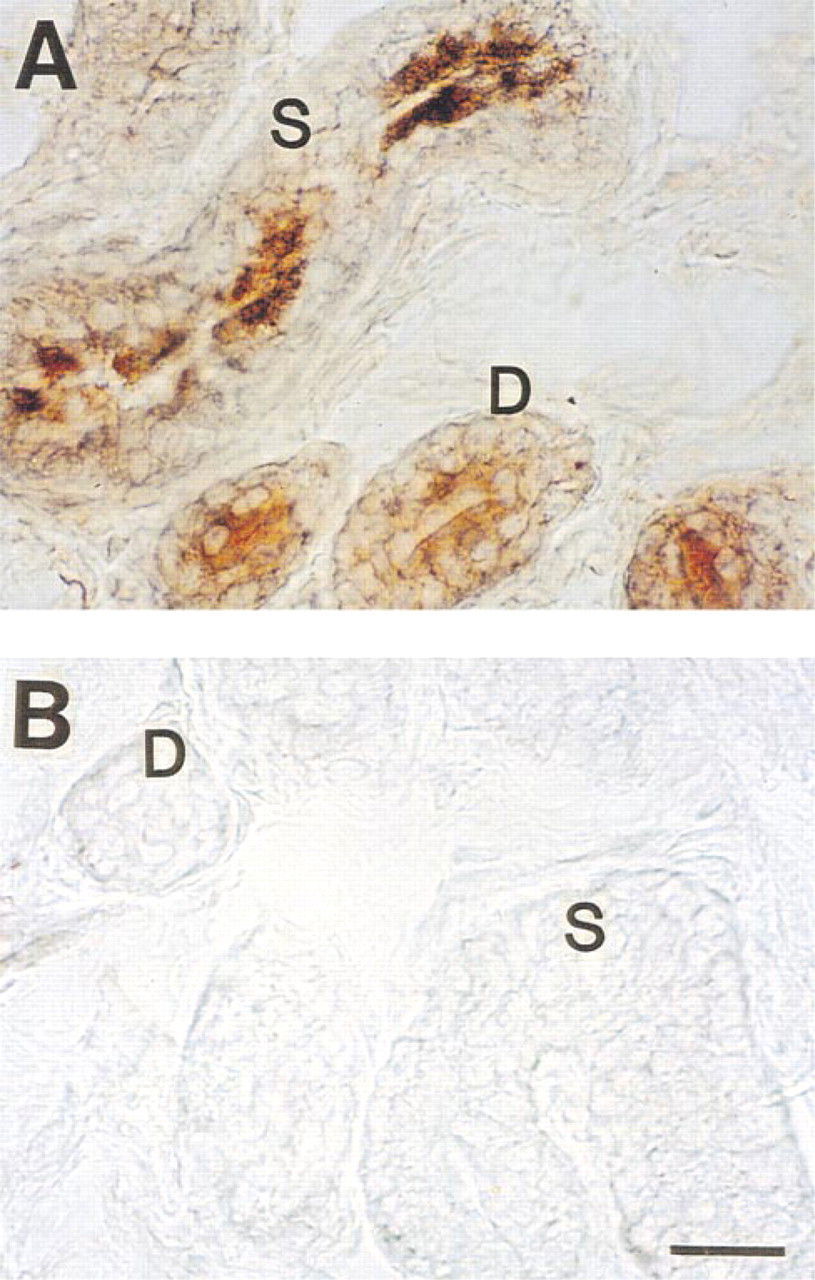
Effect of peptide blocking on MAb WC staining of CFTR C-terminus in cryosections of the sweat gland. (
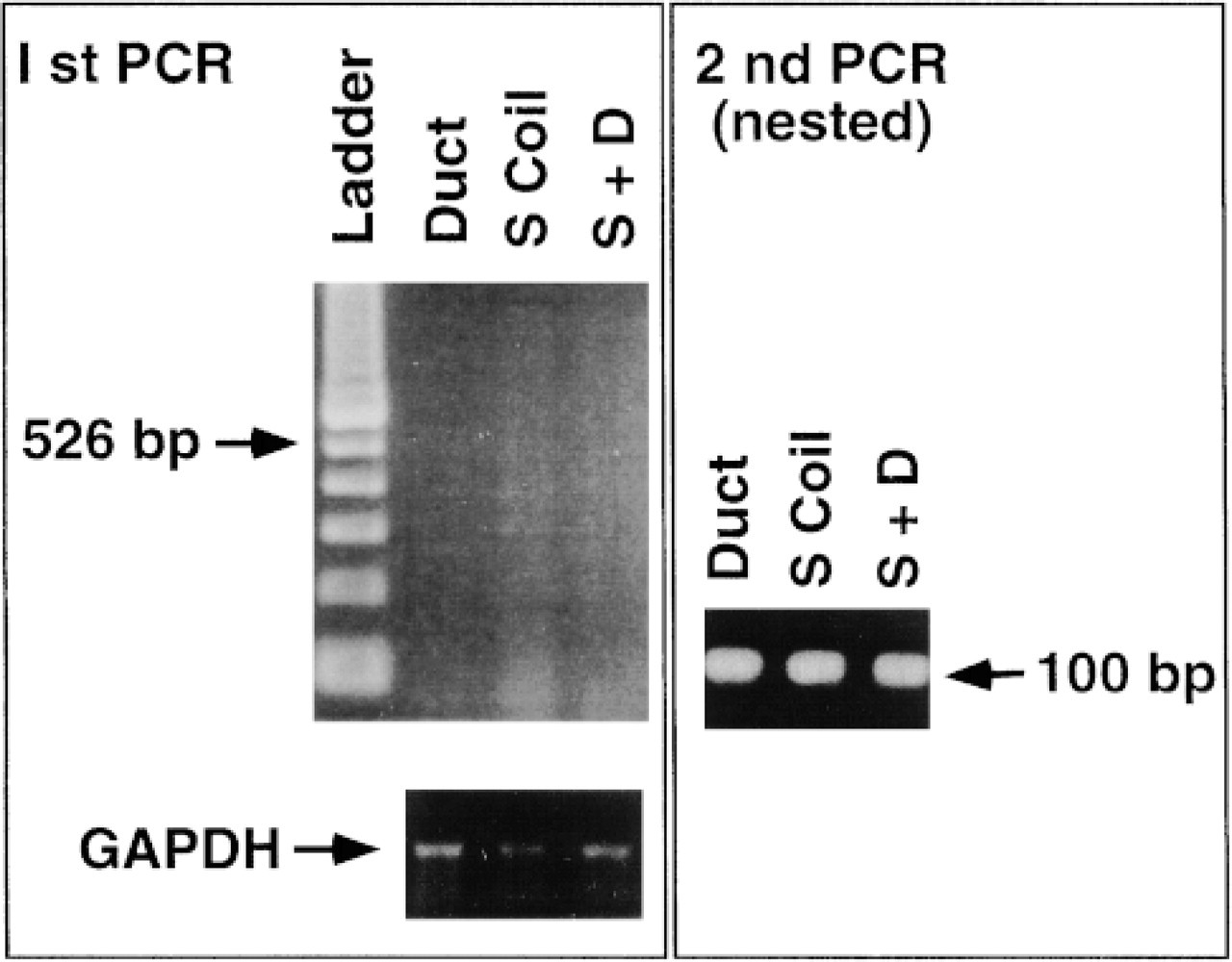
Figure 6 RT-PCR of CFTR mRNA in isolated secretory coils and ducts of human sweat glands. Secretory coils and ducts are isolated from 15 sweat glands. The first RT-PCR yielded no visible bands but the nested second PCR revealed dense bands for both the ducts and the secretory coils to a similar extent, at the predicted length of the product at 100 bp. Note that GAPDH (glyceraldehyde-3-phosphate dehydrogenase, a housekeeping gene) bands approximate the amount of original RNA isolated from the tissue.
Nested RT-PCR of CFTR in Human Sweat Glands
The first-stage RT-PCR using 15 secretory coils or ducts as the original source of mRNA did not yield a visible band (it usually takes 50-100 sweat glands to obtain a visible band; data not shown). The second nested primer RT-PCR amplified a visible band at the predicted length of 100 bp for both the duct and the secretory coil to a similar extent (Figure 6).
In Situ Localization of CFTR mRNA in Cryosections of Human Sweat Gland After 3SR
In situ localization of mRNA after 3SR is shown in Figure 7. In the secretory coil, mRNA is localized mainly in the cytoplasm near the secretory coil lumen, suggesting that the dark cells (large open arrows) and perhaps also the luminal side of clear cell cytoplasm is where mRNA is localized. In the duct, mRNA is present in both the luminal and basal ductal cells (slender arrows in Figure 7).
Discussion
Immunoreactive CFTR protein has been consistently demonstrated by three groups of investigators (Cohn et al. 1991; Crawford et al. 1991; Kartner et al. 1992) in the sweat duct, but not in the secretory coils, of non-CF sweat glands. Cryosections were used in all these studies. Kartner et al. (1992) also noted that CFTR immunoreactivity was abolished in conventional formalin-fixed, paraffin-embedded sections. The investigators also noted that only a few MAbs successfully stained CFTR proteins among several MAbs for different epitopes that were tested. Most authors used MAbs against epitopes in R-domain, NBF 1 or NBF 2, although Cohn et al. (1991) also used an MAb against C-terminal amino acids (CFTR amino acids 1468-1480).
Eccrine sweat secretion is predominantly cholinergic in nature, and cytosolic Ca2+ plays a key role in regulating stimulus secretion coupling (Sato et al. 1989), suggesting that Cl- channels are primarily regulated by cytosolic Ca2+. This raises the possibility that CFTR is not expressed or is expressed less prominently in the secretory coil, as suggested by these earlier observations (Cohn et al. 1991; Crawford et al. 1991; Kartner et al. 1992). Nevertheless, β-adrenergic stimulation and/or cAMP-elevating agents also induce sweat secretion (although to a lesser extent than does cholinergic stimulation) in control human sweat glands but not in CF sweat glands (Sato and Sato 1984). We therefore hypothesized that cAMP-dependent Cl- channels may also be involved in the control of sweat glands, which may be abnormal in CF. Furthermore, cAMP-stimulated whole-cell Cl- current was observed in isolated simian clear cells, a surrogate model for the human sweat glands (Sato et al. 1993). Therefore, it is of importance to resolve the puzzle that although CFTR is expected to be present and functionally active in clear cells, it could not be convincingly demonstrated histochemically by earlier investigators. Furthermore, the expression of CRTR gene has never been documented in freshly isolated sweat secretory cells.
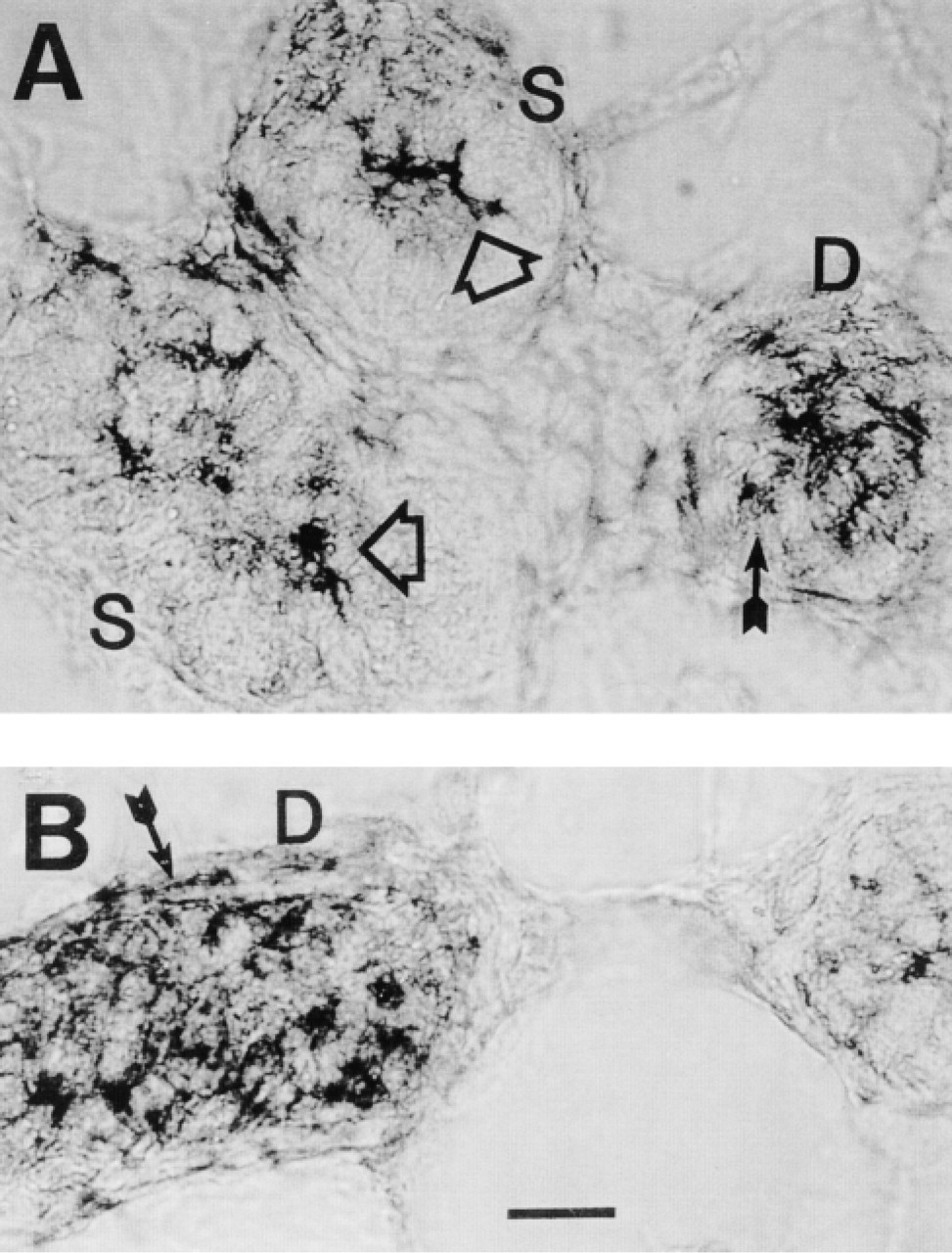
In situ localization of CFTR mRNA in cryosections of human sweat gland after 3SR. (
As shown in the present study, the secretory coil is as densely stained as the duct with the MAbs used (one MAb for the R-domain and two MAbs for the C-terminus) in cryosections, Pen-Fix-fixed paraffin-embedded sections, and conventional formalin-fixed, paraffin-embedded sections after antigen retrieval. The observed immunohistochemical staining may be specific for CFTR because the staining by both MAbs against the C-terminus was completely blocked by the C-terminus peptide (R-domain peptides were not available for this study). An alternative explanation may be that some immunoreactivity to MAbs observed is due to degradation products of CFTR (Kartner et al. 1992), due to shared epitopes expressed by unknown proteins, or due to nonspecific staining, if any. We therefore conclude that immunoreactive CFTR is present equally in both the duct and the secretory coil. This notion is further supported by demonstration of CFTR mRNA in both the duct and the secretory coil using nested RT-PCR. The fact that dark cells took up a dense staining with MAb WC but not with MAb GC is difficult to interpret because both MAb WC and MAb GC are presumed to recognize the same epitope (i.e., C-terminal four amino acids; Genzyme product insert) and that the staining by both MAbs is equally well abolished by blocking with a C-terminal peptide. Thus it is safe to conclude that all cell types (except perhaps myoepithelial cells) express immunoreactive CRTR proteins (but see above; and Kartner et al. 1992). Such a conclusion is further supported by our in situ hybridization for CFTR mRNA after 3SR amplification that demonstrates the localization of mRNA in clear and dark cells as well as basal and luminal duct cells. Accumulation of CFTR mRNA near the luminal membrane of both clear and dark cells (Figure 7) (note that clear cells also partially form the coil lumen, although dark cells predominantly line the coil lumen; see Figures 1, 2, and 4) may be instrumental for the cell to provide the newly synthesized CFTR to the luminal cell membrane where CFTR is most likely located (Sato et al. 1989). Nevertheless, the presence of immunoreactive CFTR throughout the cell cytoplasm in both secretory coil cells begs the question as to its synthesis, storage, turnover, and/or degradation. It is also of interest to observe that sweat protein and/or glandular debris secreted into the lumen is also stained by both MAbs, indicating that effete CFTR is ultimately degraded and extruded from the cell cytoplasm into sweat in the coil lumen. The possibility that CFTR protein extruded into the secretory coil lumen is taken up by the duct cells and ritualized as a CFTR Cl-channel in the duct is unlikely because the duct has its own CFTR mRNA.
The present study raises a number of questions that are of interest for future studies. For example, what is the function of CFTR in the dark cell? Because the dark cell does not appear to have CFTR channel activity in simian dark cells (Sato et al. 1993), does CFTR have an unknown function in addition to its traditional role, i.e., cAMP-dependent Cl- channels? CFTR is reported to be involved not only in Cl transport but in nonchannel function such as epithelial cell receptors for Pseudomonas aeruginosa, for its clearance from the lung (Pier et al. 1997). Discrete staining of dark cells with MAb WC may also be instrumental in that such an MAb may be useful as a marker for dark cells because the function, ontogeny, and differentiation of dark cells in vivo and in vitro is totally unknown. The disparate behavior of MAb WC and MAb GZ for recognition of CFTR in dark cells deserves further study.
Footnotes
Acknowledgments
Supported in part by NIH grants DK 27857 and AR 25339.
Thanks are also due to Dr M. Welsh and his associates for the gift of MAb WC and its blocking peptide. Dr Charles Link (director, Human Gene Therapy Research Institute) proofread the manuscript. Dr Gyula Soos was also involved in the preliminary stage of studies on antigen retrieval and in situ 3SR.