
Abstract
Objective:
Previous randomised, double-blind, placebo-controlled studies have shown that Kava (a South Pacific medicinal plant) reduced anxiety during short-term administration. The objective of this randomised, double-blind, placebo-controlled study was to perform a larger, longer-term trial assessing the efficacy and safety of Kava in the treatment of generalised anxiety disorder and to determine whether gamma-aminobutyric acid transporter (SLC6A1) single-nucleotide polymorphisms were moderators of response.
Methods:
The trial was a phase III, multi-site, two-arm, 16-week, randomised, double-blind, placebo-controlled study investigating an aqueous extract of dried Kava root administered twice per day in tablet form (standardised to 120 mg of kavalactones twice/day) in 171 currently non-medicated anxious participants with diagnosed generalised anxiety disorder. The trial took place in Australia.
Results:
An analysis of 171 participants revealed a non-significant difference in anxiety reduction between the Kava and placebo groups (a relative reduction favouring placebo of 1.37 points; p = 0.25). At the conclusion of the controlled phase, 17.4% of the Kava group were classified as remitted (Hamilton Anxiety Rating Scale score < 7) compared to 23.8% of the placebo group (p = 0.46). No SLC6A1 polymorphisms were associated with treatment response, while carriers of the rs2601126 T allele preferentially respond to placebo (p = 0.006). Kava was well tolerated aside from poorer memory (Kava = 36 vs placebo = 23; p = 0.044) and tremor/shakiness (Kava = 36 vs placebo = 23; p = 0.024) occurring more frequently in the Kava group. Liver function test abnormalities were significantly more frequent in the Kava group, although no participant met criteria for herb-induced hepatic injury.
Conclusion:
While research has generally supported Kava in non-clinical populations (potentially for more ‘situational’ anxiety as a short-term anxiolytic), this particular extract was not effective for diagnosed generalised anxiety disorder.
Introduction
Anxiety disorders represent the most prevalent cluster of psychiatric conditions, being responsible for marked societal suffering and economic cost (Baxter et al., 2012). Generalised anxiety disorder (GAD) is a chronic, debilitating psychiatric disorder, associated with significantly impaired occupational and social functioning (Ruscio et al., 2017). Present pharmacotherapies used to treat GAD, while moderately efficacious, may not always lead to patient remission. A review of the treatment guidelines involving pharmacological therapy has revealed that one third of the patients are expected not to respond to first-line treatment, with another half also being potentially unresponsive to second-line treatment (Bereza et al., 2012). Clearly, more treatment options are needed for the treatment of GAD. A particular approach with significant public interest involves the use of nutrient-based (nutraceuticals) or plant-based (phytoceuticals) medicines (Barić et al., 2018). One such candidate is Kava (Piper methysticum), a South Pacific plant medicine with traditional cultural and recreational uses and extensive modern use as a calmant (e.g. in popular ‘Kava bars’ in the United States) (Sarris et al., 2011).
Numerous preclinical studies indicate that a class of the bioactive constituents, known as kavalactones, impart an array of neurobiological effects, with anxiolysis occurring primarily via modulation of the gamma-aminobutyric acid (GABA) pathway (Sarris et al., 2011). Specific actions include reduced excitatory neurotransmitter release due to blockade of calcium ion channels (Martin et al., 2002; Walden et al., 1997), enhanced ligand binding to GABA type A receptors (Jussofie et al., 1994), blockade of voltage-gated sodium ion channels (Gleitz et al., 1995; Magura et al., 1997), reversible inhibition of monoamine oxidase B (Uebelhack et al., 1998) and reduced neuronal reuptake of dopamine (Baum et al., 1998) and noradrenaline (Seitz et al., 1997). With respect to clinical evidence, our previous systematic review involving a pooled analysis of six studies using Kava versus placebo in the treatment of anxiety symptoms revealed a significant effect in favour of Kava on the Hamilton Anxiety Rating Scale (HAMA), with an effect size (Cohen’s d) of 1.1 (Sarris et al., 2011). While being potentially efficacious for anxiety, the World Health Organization–commissioned report in 2006 assessing Kava products called for clinical trials using water-based extractions in order to firmly establish Kava’s efficacy and safety, due to concerns at the time about liver toxicity related to ethanolic and acetonic extracts (Coulter, 2007). Subsequently, it should be noted that negative effects on the liver were not substantiated by the German Regulatory Authority (Kuchta et al., 2015).
Responding to the call for more research, our team has conducted a series of clinical trials. Initially, we examined the short-term use of a standardised water-extracted Kava (five tablets [a total of 250 mg of kavalactones per day]) in a randomised, double-blind, placebo-controlled (RCT), balanced, crossover trial (n = 60) (Sarris et al., 2009). One week of prescribed Kava significantly reduced participants’ anxiety compared to placebo on the HAMA (Cohen’s d = 2.24, p < 0.0001). Next, we built upon these initial findings by conducting a 6-week RCT (n = 75) in which adults with GAD and no comorbid depression were given one tablet of water-extracted Kava twice per day (titrated to two tablets twice per day in non-response [to a maximum of 240 mg of kavalactones per day]) or matching placebo (Sarris et al., 2013). The results revealed a significant reduction in anxiety in favour of the Kava group compared to placebo (p = 0.046), with a medium effect size (d = 0.63). We also found that polymorphisms within the SLC6A1 gene (a key gene coding for the GABA transporter protein) may have moderated the response to Kava (i.e. rs2601126 T alleles, p = 0.021, or rs2697153 A alleles, p = 0.046). In both studies, no differential abnormalities between treatment groups on liver function tests (LFTs) were revealed.
Based on these compelling pilot data, we sought to confirm the effectiveness and safety of Kava as a potential first-line pharmacological approach to treating GAD via a longer-term, multi-site RCT, with a larger sample. We also sought to replicate the association between GABA transporter polymorphisms and response to Kava treatment.
Materials and methods
Study design and procedure overview
The design of the study was a phase III, multi-site, two-arm, 16-week RCT using a standardised pharmaceutical-grade water extract of Kava (240 mg of kavalactones per day, derived from the dried roots of a ‘noble’ Borogu cultivar sourced from Vanuatu) or matching placebo in currently anxious participants with diagnosed GAD (for further information, see protocol paper; Savage et al., 2015). Participants were required to attend seven visits at the trial sites at weeks 0 [baseline], 2, 4, 8, 12 and 16 (with a 2-week placebo-runout follow-up visit occurring after week 16). The trial sites were at the Centre for Human Psychopharmacology (Swinburne University of Technology), Hawthorn, Melbourne, Australia, and Academic Discipline of Psychiatry (The University of Queensland) based at The Royal Brisbane and Women’s Hospital, Herston, Brisbane, Australia. Recruitment occurred between October 2015 and January 2018. The study had ethical clearance (Alfred Hospital – No. HREC137/14; University of Queensland – No. HREC2014000876; Swinburne University – No. HREC2014/204) and is registered on ClinicalTrials.gov (protocol no. NCT02219880). All participants provided written informed consent prior to taking part in the trial. Participants were reimbursed (in voucher form) for their travel expenses at a total value of AU$200 for participants who completed the entire study. See Supplementary Material for more details on the procedure.
Screening and outcome measures
The Mini-International Neuropsychiatric Interview (MINI 6.0; Sheehan et al., 1998) was used to diagnose Diagnostic and Statistical Manual of Mental Disorders (5th ed.; DSM-5) GAD as well as to screen for other anxiety disorders, major depressive disorder (MDD), psychotic disorders, bipolar disorder and substance/alcohol dependency or abuse.
The primary outcome was anxiety symptom reduction as measured by the HAMA (Hamilton, 1959) at each time point. Secondary outcomes assessed at every visit were as follows: self-reported anxiety levels on the Beck Anxiety Inventory (BAI; Beck et al., 1988); worry, via the Penn State Worry Questionnaire (PSWQ; Meyer et al., 1990); mood on the Montgomery–Asberg Depression Rating Scale (MADRS; Montgomery and Asberg, 1979); quality of life via the World Health Organization Quality of Life – BREF (WHOQOL-BREF; Skevington et al., 2004); perceived stress via the Kessler Psychological Distress Scale (K-10; Kessler et al., 2002); sexual functioning via the Arizona Sexual Experiences Scale (ASEX; McGahuey et al., 2000) and sleep disturbance on the Insomnia Severity Index (ISI; Morin et al., 2011). Side effects were assessed using the Systematic Assessment for Treatment Emergent Effects (SAFTEE) questionnaire (Levine and Schooler, 1986).
Participants (inclusion and exclusion criteria)
To be eligible for the study, participants were required to be between the ages of 18 and 70 years, meet DSM-5 diagnostic criteria for GAD (based on the MINI 6.0 structured interview) and present with moderate levels of anxiety (HAMA ⩾ 18) at the time of study entry. Other inclusion criteria were fluency in written and spoken English and ability to provide written informed consent. The main exclusion criteria were a primary psychiatric diagnosis other than GAD; moderate to severe depressive symptoms (MADRS score of ⩾18) at time of study entry (or suicidality); pertinent current medications (e.g. antidepressants, mood stabilisers or antipsychotics, or regular use of benzodiazepines or opioid-based analgesics); previous intolerance to Kava; three or more failed trials of pharmacotherapy for the current GAD episode; recently commenced psychotherapy; known or suspected clinically unstable systemic medical disorder or diagnosed hepatobiliary disease/inflammation (see Supplementary Material for full exclusion criteria).
Treatment interventions, blinding and randomisation
Kava and placebo were supplied in tablet form and were manufactured to be identical in colour, size, shape and odour (a porous sachet containing milled Kava dried root powder was added to all treatment bottles to further enhance blinding). Tablets were manufactured from a hot aqueous extract (drug extract ratio 7:1) of the dried roots of a ‘noble’ Borogu cultivar, sourced from Vanuatu. Each tablet was standardised to 60 mg total kavalactones per tablet (kavain: 29.23%, dihydrokavain: 24.42%, trans-yangonin: 13.88%, cis-yangonin: 0.66%, des-methoxy yangonin: 10.21%, dihydromethysticin: 10.31%, methysticin: 11.30%, validated by high-performance liquid chromatography), with a calculated chalcone (flavokavain) level of 2.62 mg per tablet (equivalent to 1.23 mg flavokavain A and 1.39 mg of flavokavain B). This reflected a high-quality Kava chemotype (Teschke et al., 2011). Both active and placebo tablets were manufactured by Integria Healthcare (Australia) Pty Ltd in accordance with pharmaceutical Good Manufacturing Practice. The manufactured study tablets were tested for contaminants by an external laboratory and met all government-required specifications for pesticides (United States Pharmacopoeia [USP]), solvent residues (USP), heavy metals and microbial counts.
The randomisation of treatment codes was performed by a disinterested third party not involved in the trial using computerised permuted 3 × 2 block randomisation, for example, ABAAABABBAB. Participants were allocated a treatment code sequentially by a researcher blinded to the randomisation schedule. Both the researchers and participants were fully blinded to group allocation.
Biomarker analyses
LFT assessment was performed by Australian Clinical Labs (Clayton South, Victoria). The panel of LFTs included total protein, albumin, alkaline phosphatase (ALP), total bilirubin, gamma-glutamyl transferase (GGT), aspartate aminotransferase (AST), alanine aminotransferase (ALT) and globulin. LFT abnormalities were defined as any deviation from normal values (see Table 2 for thresholds). The following criteria were used to flag a potential herb-induced liver injury (HILI): (1) ALT ⩾ 5× upper limit of normal (ULN) or (2) ALP ⩾ 2× ULN (Danan and Teschke, 2016). These criteria paralleled those proposed for a drug-induced liver injury by a working group of international experts (Aithal et al., 2011).
Three single-nucleotide polymorphisms (SNPs; rs2601126, rs2697153 and rs9990174) within SLC6A1 were genotyped. DNA was extracted from whole blood utilising Qiagen QIAamp mini-columns according to manufacturer instructions. SNPs were identified using single-base extension assays and analysed on the Sequenom MassARRAY at Australian Clinical Laboratories.
Statistical analysis
A recruitment target of 210 participants (105 per arm) was outlined in the protocol, allowing the detection of a small/medium effect size (Cohen’s f) of 0.15, with 80% power and a 5% error rate. This was based on an analysis of variance (ANOVA) with six time points and a correlation among repeated measures. The achieved sample size of 171 participants was below this target. Nevertheless, this sample size continued to provide power sufficient for the detection of a small/medium effect size of 0.17, equating roughly to a 2.5 point between-group difference in scores on the primary outcome (HAMA).
An analysis of data was conducted blinded to group allocations. Sociodemographic and clinical characteristics as well as frequencies of adverse events and liver abnormalities were tested for inter-group differences using t tests for continuous variables and chi-square tests for categorical variables. The Mann–Whitney U test was used when assumptions for parametric tests were violated. Pertinent sociodemographic and clinical characteristics were tested for association with the outcome (change in HAMA score) using ANOVA models. ‘Needing medication to function’ (WHOQOL-BREF), concurrent use of a non-psychiatric medication and history of a depressive episode all predicted change in HAMA score (p < 0.10). These variables, along with treatment site, were retained for use as covariates in subsequent, primary (HAMA) models. Analyses of primary and secondary outcomes were undertaken utilising linear mixed-effects models (LMMs), with the outcome of interest the group × time interaction (representing a differential response gradient between groups which signified a treatment effect). A random intercept and slope model was utilised in each LMM excluding the PSWQ (random intercept only), as this matched the visual structure of the data and was supported by significance tests. An autoregressive covariance structure was also utilised in each model. Significance was set at p < 0.05. Effect sizes (Cohen’s d) were calculated based on endpoint HAMA scores, as baseline HAMA scores were effectively equivalent. Finally, an HAMA score of <7 at study completion (week 16) was selected as a marker of remission, while response was defined as a reduction in the HAMA score of more than 50%. Statistical analyses were performed using the Statistical Package for the Social Sciences (SPSS) software version 25 (IBM SPSS). For details on the analysis methods for LFTs and SNPs, see Supplementary Material.
Results
Recruitment and participant characteristics
A total of 230 individuals attended a baseline visit and were assessed for eligibility, with 59 being ineligible for inclusion (see Figure 1 for participant flow through the trial). A total of 171 participants met inclusion criteria and gave consent to participate in the 18-week study. Each of these 171 participants were analysed in an intention-to-treat (ITT) analysis, with no significant difference in drop-out rates between groups (39 drop-outs in placebo vs 40 drop-outs in Kava), χ2(1, 171) = 0.007, p = 0.93. Sociodemographic and clinical features of the sample are displayed in Table 1. There were no significant between-group differences in any of the assessed variables (all p values > 0.05).
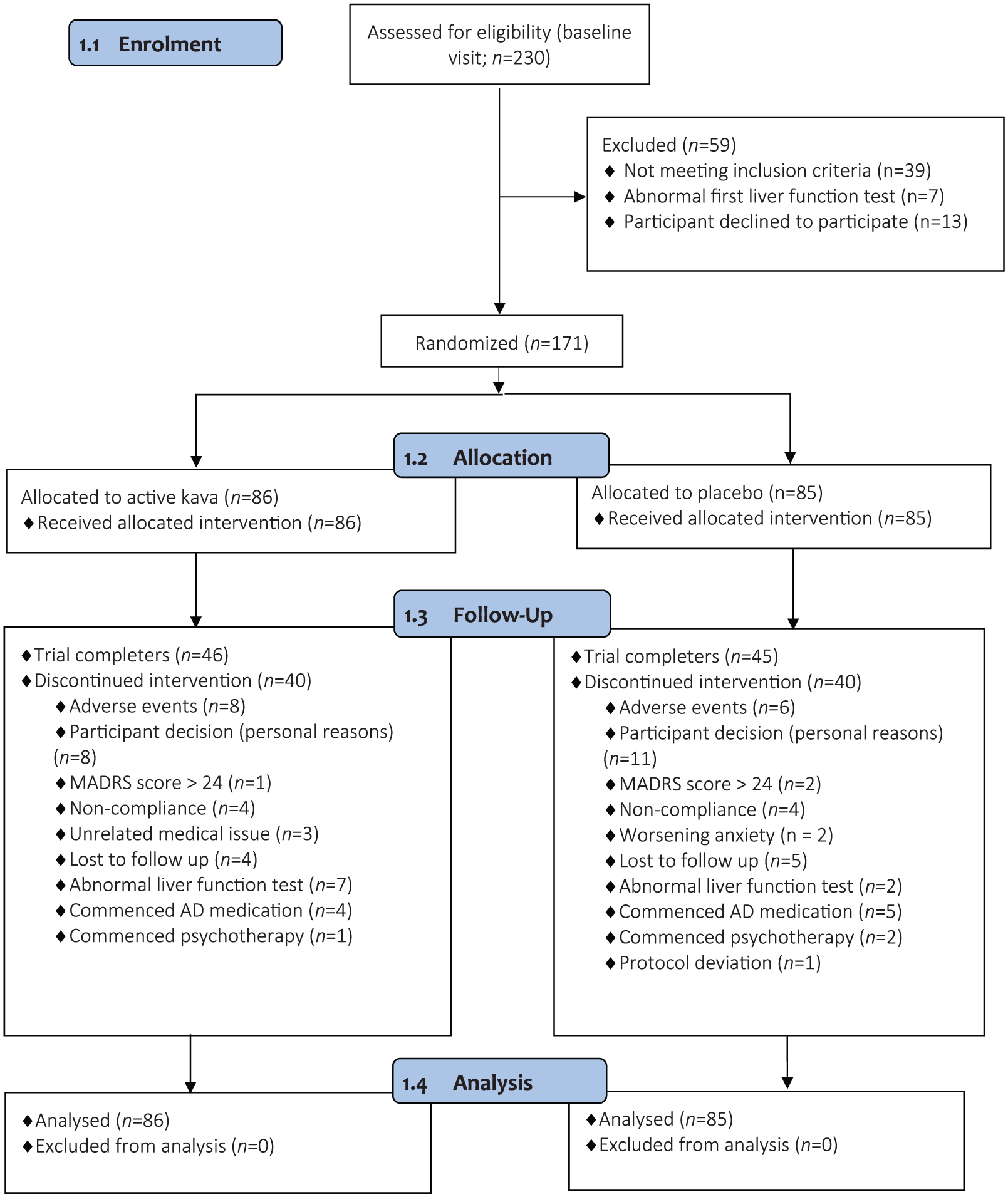
CONSORT flow diagram.
Sociodemographic and clinical characteristics. a
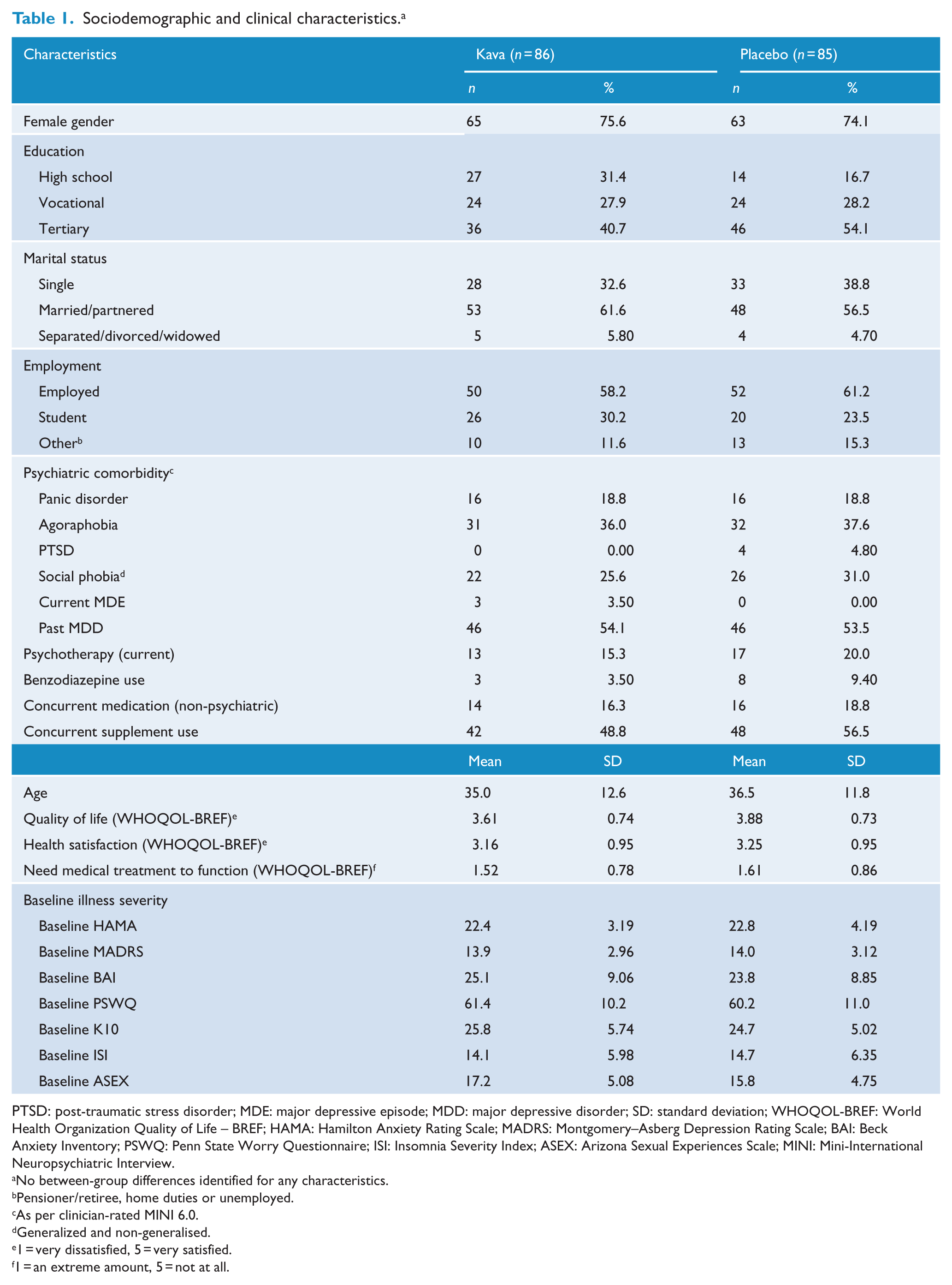
PTSD: post-traumatic stress disorder; MDE: major depressive episode; MDD: major depressive disorder; SD: standard deviation; WHOQOL-BREF: World Health Organization Quality of Life – BREF; HAMA: Hamilton Anxiety Rating Scale; MADRS: Montgomery–Asberg Depression Rating Scale; BAI: Beck Anxiety Inventory; PSWQ: Penn State Worry Questionnaire; ISI: Insomnia Severity Index; ASEX: Arizona Sexual Experiences Scale; MINI: Mini-International Neuropsychiatric Interview.
No between-group differences identified for any characteristics.
Pensioner/retiree, home duties or unemployed.
As per clinician-rated MINI 6.0.
Generalized and non-generalised.
1 = very dissatisfied, 5 = very satisfied.
1 = an extreme amount, 5 = not at all.
Anxiety
Estimated marginal means derived from the primary LMM revealed a decrease in HAMA score, from baseline to week 16, of 7.56 points in the Kava group and 8.93 in the placebo group (relative reduction favouring placebo of 1.37 points; see Figure 2). The primary LMM revealed no significant effect of treatment on the HAMA (group × time interaction), F(5, 428) = 1.34, p = 0.25. This equated to a very small effect size of 0.10 in favour of placebo in reducing anxiety. Neither sex, age, baseline anxiety severity, presence of psychiatric comorbidity, current psychotherapy or use of benzodiazepines moderated the relationship between treatment arm and outcome on the HAMA (group × time × covariate interaction: all p values > 0.05). Treatment response (HAMA reduction > 50%) was present in 43% of participants within the placebo group and 28% within the Kava group, which was not significantly different between groups, χ2(1, 88) = 2.05, p = 0.15. Remission (HAMA < 7 at study completion) was reached by 24% of participants in the placebo group and 17% of participants in the Kava group, which was similarly non-significant between groups, χ2(1, 88) = 0.556, p = 0.46.
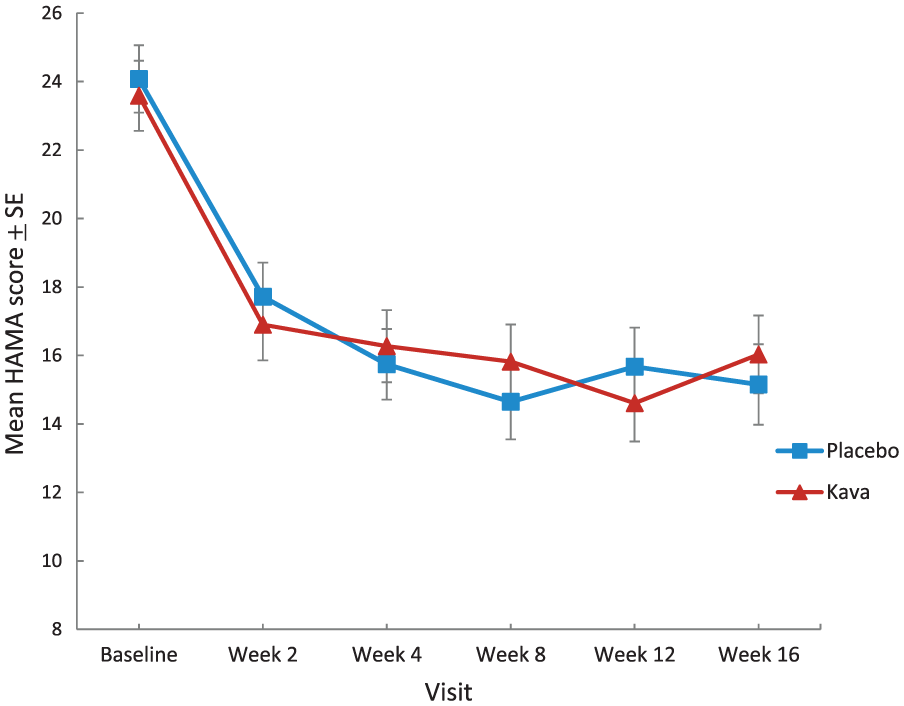
Reduction in anxiety on the HAMA.
Secondary analyses
A marginally non-significant treatment effect (group × time interaction) was noted on the PSWQ, F(5, 420) = 2.23, p = 0.051, in favour of placebo reducing ‘worry’. Post hoc tests revealed a significant difference in the mean PSWQ score (4.20 points) at week 16, in favour of placebo, F(1, 386) = 4.04, p = 0.045. No significant effect of treatment was noted on the MADRS, F(5, 449) = 0.299, p = 0.91; K10, F(5, 420) = 1.40, p = 0.23; BAI, F(5, 341) = 0.948, p = 0.46; ISI, F(5, 357) = 1.02, p = 0.41; or ASEX, F(5, 373) = 0.574, p = 0.72.
Genetic correlates
Minor allele frequencies of the three investigated GABA transporter SNPs (rs2601126C>T, rs2697153G>A and rs9990174G>T) were 0.36(T), 0.42(G) and 0.29(T), respectively. Each of the SNPs was in Hardy–Weinberg equilibrium (HWE; all p values > 0.05). Two of the SNPs (rs2601126 and rs2697152) were in strong linkage disequilibrium (R2 = 0.38, D′ = 0.97) and were analysed as a haplotype block (see Supplementary Material). Inspection of Akaike information criterion values revealed that a dominant mode of inheritance produced the best model fit for rs2601126 and rs2697152, while a recessive mode of inheritance was appropriate for rs9990174.
A significant group × time × rs2601126 interaction was noted on the HAMA, F(5, 371) = 3.34, p = 0.006, indicating that rs2601126 moderated the relationship between group and reduction in anxiety symptoms. In particular, T allele carriers were more responsive to placebo, relative to treatment (see Figure 3). Further genomic data and the haplotype analysis are available in Supplementary Material.
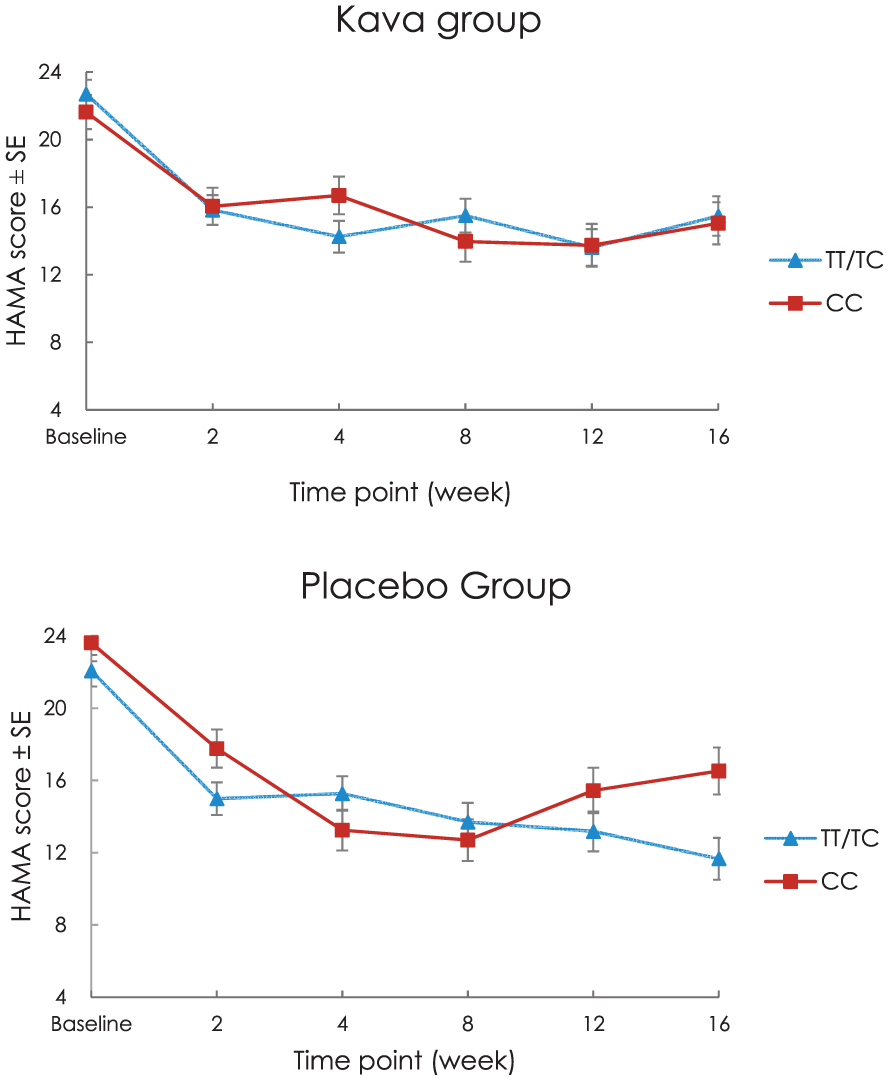
Effect of rs2601126 genotype on HAMA response by group.
Safety evaluation
Two serious adverse events were recorded in the trial, both in the placebo group. Each serious adverse event was deemed unrelated to study treatment by the study medical investigators. There were no differences in total adverse events self-reported on the SAFTEE (U = 3115, Z = –0.289, p = 0.77). Differences were evident, however, for symptoms of ‘poor memory’ (Kava = 36 vs placebo = 23) and ‘tremor or shakiness’ (Kava = 13 vs placebo = 4), which were significantly more frequent in the Kava group, χ2(1, 160) = 4.04, p = 0.044, and χ2(1, 160) = 5.08, p = 0.024, respectively. No withdrawal effects were evident for Kava participants on health domains such as neurological, digestive, respiratory or cardiovascular function.
LFTs
Over the study period, 8% of the participants (n = 7) in the Kava group were withdrawn due to LFT abnormalities (as a safety precaution), compared to 2% (n = 2) in the placebo group. This was non-significantly different (Fisher’s exact test, p = 0.17). After randomisation, at week 2, there were no differences in LFT abnormalities between placebo (n = 5; 6%) and Kava (n = 6; 9%), χ2(1, 147) = 0.683, p = 0.41. At week 16 (all data carried forward via an ITT analysis), significantly more post-baseline LFT abnormalities were present in the Kava group (n = 17; 24%) than the placebo group (n = 6; 8%), χ2(1, 149) = 7.13, p = 0.008. Specifically, there was a significantly higher proportion of abnormalities in GGT, AST and ALT in the Kava group (p = 0.024, p = 0.011 and p = 0.041, respectively; see Table 2). This equated to a 116% greater risk of a post-baseline LFT abnormality in the Kava group relative to placebo (risk ratio [RR]: 2.16, 95% confidence interval [CI]: [1.07, 4.37]). After excluding participants with LFT abnormalities at baseline, which were over-represented in the Kava group (10 vs 4 in the placebo group), the increased risk of a post-baseline LFT abnormality associated with Kava treatment was attenuated (RR: 1.57, 95% CI: [0.808, 3.05]). Within the Kava group, participants whose mean weekly alcohol consumption was in the top 20th percentile (>6.2 standard units) experienced LFT abnormalities at a greater frequency (n = 7; 41%) than those who consumed less alcohol (n = 10; 18%), χ2(1, 72) = 3.81, p = 0.051. By contrast, the top 20th percentile mean weekly alcohol consumption was not associated with a greater frequency of LFT abnormalities in the placebo group (n = 0; 0%). Further LFT results are available in Supplementary Material.
Liver function test scores.
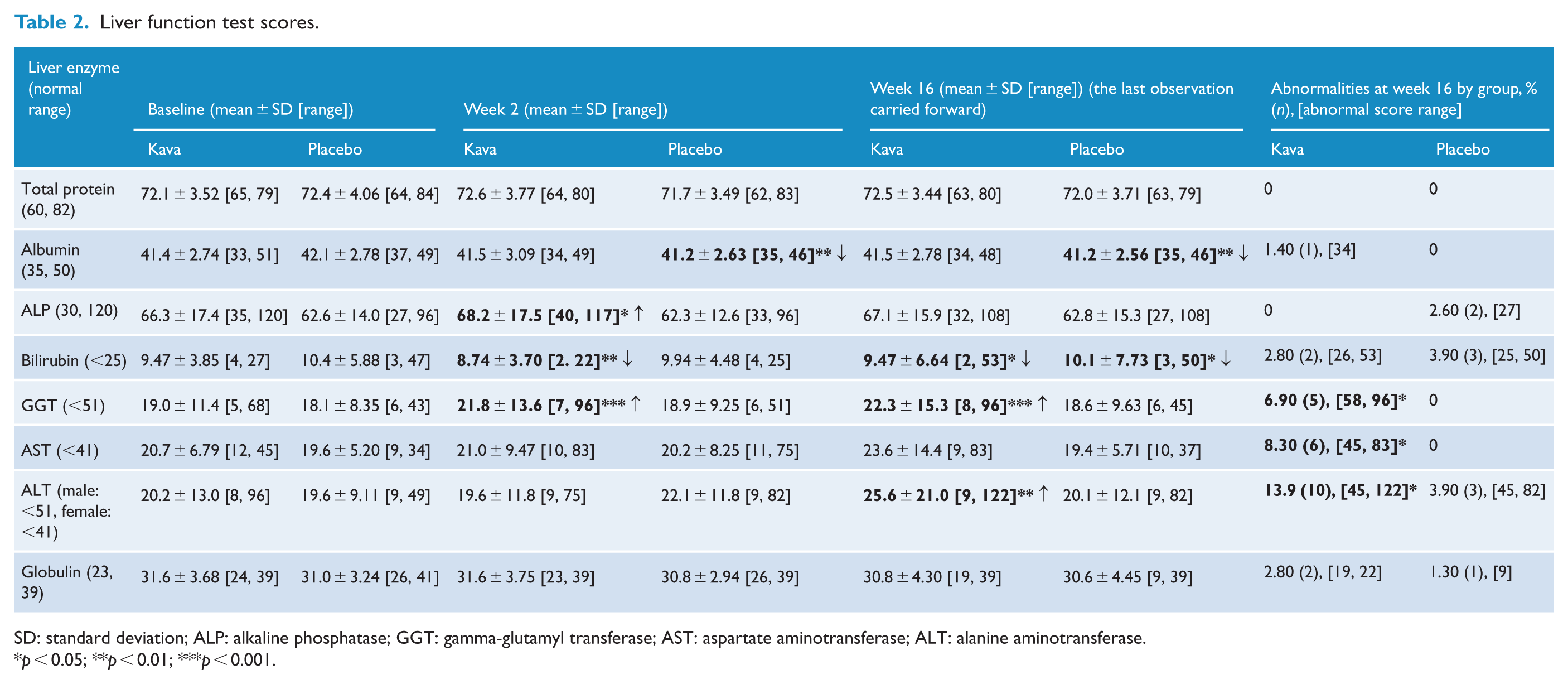
SD: standard deviation; ALP: alkaline phosphatase; GGT: gamma-glutamyl transferase; AST: aspartate aminotransferase; ALT: alanine aminotransferase.
p < 0.05; **p < 0.01; ***p < 0.001.
Discussion
This clinical trial did not replicate the positive results of our last RCT (Sarris et al., 2013), thus not confirming Kava as a first-line intervention for GAD. These data can be added to the results of a partially completed study in 2002 by Connor and Davidson (2002) who showed that Kava was less effective (again non-significantly) than placebo for treating GAD. Even in our previous positive pilot study in GAD, it should be noted that Kava achieved a modest response rate, improving outcomes in 37% of those in the treatment arm, indicating that it was not appreciably effective in the majority of the sample. In this study, the response rate was even lower, at 28%. However, while not effective for GAD, based on the accumulation of other research (and underpinning preclinical evidence), Kava still has evidentiary support for non-clinical generalised anxiety.
It should however be noted that pharmacological treatment of GAD is in general challenging. Although most pharmaceutical treatments tend to outperform placebo (Baldwin et al., 2011; Slee et al., 2019), a recent network meta-analysis reported that each of the investigated therapies (excluding those with inadequate sample sizes) produced mean differential reductions of less than four points on the HAMA (Slee et al., 2019). These differences suggest that substantial, clinically important reductions in GAD symptomology are difficult to achieve. This is also further compounded by the tolerability issues of some of these pharmacological agents compared to placebo (Slee et al., 2019).
The pharmacogenetic results from this analysis also did not support previous findings related to genetic variation within the GABA transporter gene (SLC6A1) of our previous study (Sarris et al., 2013). In the previous RCT, we reported a positive correlation between the number of rs2601126 T alleles and rs2697153 A alleles and response to treatment. In this study, however, there was no clear association between SLC6A1 SNPs and treatment response in the Kava group. Response was however greater on the HAMA, BAI and ISI, for T allele carriers of the rs2601126 SNP within the placebo group. While being a puzzling result, it may however be important for the emerging field of ‘placebomics’.
With respect to any interaction with liver function, our previous RCT for GAD found no effect of Kava treatment on LFT results (Sarris et al., 2013). This study, however, revealed that abnormal results occurred for more participants in the Kava group than in the placebo group, particularly mild elevations in GGT, AST and ALT levels. Risk of LFT abnormalities during the Kava treatment was heightened, and furthermore so in males and those with higher alcohol consumption (although sample sizes were small for these analyses). It is important to note that, although LFT abnormalities were more frequent in the Kava group, no participant met criteria for an HILI, and such LFT changes may potentially be viewed as a liver adaptation response (Danan and Teschke, 2016). Finally, aside from these findings, Kava was well tolerated with the caveat that ‘poor memory’ and ‘tremor or shakiness’ were more frequent compared to placebo.
Our study has several strengths, including a large sample size, a longer period of administration, a standardised form of Kava, rigorous blinding and placebo control as well as strict exclusion criteria. We do however recognise an uncontrollable limitation which concerned the rate of participant drop-out, which is less prevalent in studies of psychological techniques in GAD (rate of around 15–20%; Gersh et al., 2017). This was however not different between groups and was deemed to be due to a range of factors which primarily concerned personal reasons, adverse events and medication changes (which are more likely to occur in longer studies such as this 18-week clinical trial). We also accept that other Kava cultivars may be more effective for the treatment of GAD (and note that the cultivar-specific raw material used in this clinical trial was different from our previous positive clinical trials, which used a blend of high-grade Vanuatu Kava). Finally, it is possible that a higher dose of Kava is needed to be effective in treating GAD; however, it is noted that recommendations exist in most jurisdictions to consume no more than 240 or 250 mg of kavalactones per day.
Conclusion
While extant data show that standardised Kava (containing either 120, 240 or 250 mg of kavalactones) is an effective short-term treatment for generalised and, potentially more so, ‘situational’ anxiety, the results of our study indicate that it is not effective as a psychotropic medication for diagnosed GAD. Aside from Kava’s valued social, cultural and recreational use, based on our findings and previous data, the plant’s therapeutic application may more appropriately extend to its use as an anxiolytic prior to a potential situational anxiogenic event, or for intermittent use in the additional management of non-clinical anxiety and stress.
Supplemental Material
Supplementary_Data – Supplemental material for Kava for generalised anxiety disorder: A 16-week double-blind, randomised, placebo-controlled study
Supplemental material, Supplementary_Data for Kava for generalised anxiety disorder: A 16-week double-blind, randomised, placebo-controlled study by Jerome Sarris, Gerard J Byrne, Chad A Bousman, Lachlan Cribb, Karen M Savage, Oliver Holmes, Jenifer Murphy, Patricia Macdonald, Anika Short, Sonia Nazareth, Emma Jennings, Stuart Thomas, Edward Ogden, Suneel Chamoli, Andrew Scholey and Con Stough in Australian & New Zealand Journal of Psychiatry
Supplemental Material
Supplementary_figure_1_ANZJP – Supplemental material for Kava for generalised anxiety disorder: A 16-week double-blind, randomised, placebo-controlled study
Supplemental material, Supplementary_figure_1_ANZJP for Kava for generalised anxiety disorder: A 16-week double-blind, randomised, placebo-controlled study by Jerome Sarris, Gerard J Byrne, Chad A Bousman, Lachlan Cribb, Karen M Savage, Oliver Holmes, Jenifer Murphy, Patricia Macdonald, Anika Short, Sonia Nazareth, Emma Jennings, Stuart R Thomas, Edward Ogden, Suneel Chamoli, Andrew Scholey and Con Stough in Australian & New Zealand Journal of Psychiatry
Footnotes
Acknowledgements
Sincere thanks are extended to the clinical trial participants.
Declaration of Conflicting Interests
J.S. has received either presentation honoraria, travel support, clinical trial grants, or independent consultancy payments from companies which sell Kava products (i.e. Integria Healthcare & MediHerb, Taki Mai, Fiji Kava, FIT-BioCeuticals and Blackmores) and also from Pfizer, Scius Health, Key Pharmaceuticals, Soho-Flordis, Healthworld, HealthEd, HealthMasters, Kantar Consulting, Grunbiotics, Research Reviews, Elsevier (book royalties), Chaminade University, International Society for Affective Disorders, Complementary Medicines Australia, SPRIM, Terry White Chemists, ANS, Australian Natural Therapies Group, Society for Medicinal Plant and Natural Product Research, Sanofi-Aventis, Omega-3 Centre, the National Health and Medical Research Council and CR Roper Fellowship. G.J.B. has received grants from Biogen, Eli Lilly and Johnson & Johnson (Janssen) for conducting clinical trials in Alzheimer’s disease and related conditions; book royalties and income from licensing of anxiety rating scales; and research grant income from the National Health and Medical Research Council and the Royal Brisbane and Women’s Hospital Foundation.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This study was funded by an Australian National Health and Medical Research Council (NHMRC) grant (No. APP1063383) and co-funded by Integria Healthcare (who were completely uninvolved in the study design, data analysis as well as write-up and editing of the results). J.S. was supported by an NHMRC Clinical Research Fellowship (No. APP1125000). The raw material used in the investigational tablets was sourced specifically for the trial and is different from that used in products currently marketed by Integria Healthcare.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.