
Abstract
Objective:
Increasing evidence suggests that inflammation and immune dysregulation play an important role in the pathogenesis of bipolar disorder. Because the brain can be affected by various autoimmune processes, it is possible that some psychiatric disorders may have an autoimmune basis.
Method:
This article reviews the literature on peripheral and central immune dysregulation and autoimmunity in bipolar disorder. The mechanisms of the innate and adaptive immune systems in the pathophysiology of bipolar disorder are explored. The clinical features and pathogenesis of neuropsychiatric systemic lupus erythematosus, anti-NMDA encephalitis, and Hashimoto’s encephalopathy are summarized.
Results:
Neuroinflammation and peripheral immune dysregulation may play a role in the pathophysiology of bipolar disorder. This involves a complex interaction between immune cells of the central nervous system and periphery resulting in cellular damage through mechanisms involving excitotoxicity, oxidative stress, and mitochondrial dysfunction. Neuropsychiatric systemic lupus erythematosus, anti-NMDA encephalitis, and Hashimoto’s encephalopathy are important differentials for a psychiatrist to consider when suspecting autoimmune encephalopathy.
Conclusions:
The link between immune dysregulation, autoimmunity, and bipolar disorder may be closer than previously thought. Psychiatrists should be vigilant for autoimmunity in presentations of bipolar disorder due to its high morbidity and therapeutic implications. Advances in neuroimaging and biomarker identification related to immune dysregulation and neuroinflammation will contribute to our knowledge of the pathophysiology of bipolar disorder.
Keywords
Introduction
The greatest mistake in the treatment of diseases is that there are physicians for the body and physicians for the soul, although the two cannot be separated (Plato)
Our understanding of the role of the immune system and autoimmunity in psychiatry is evolving. A large body of evidence points to a role for inflammatory mediators and immune dysregulation in the pathogenesis of psychiatric disorders such as schizophrenia, depression, and Alzheimer’s disease (Fan et al., 2007; Krishnadas and Cavanagh, 2012; Moscavitch et al., 2009; Zotova et al., 2010). More recently, evidence has accumulated suggesting that inflammation and immune dysregulation play an important role in neuroprogression in bipolar disorders (Berk et al., 2011).
The link between autoimmunity and psychiatric symptoms has long been recognized, and W. Osler offered a description of psychosis in systemic lupus erythematosus (SLE) in 1895. In neurology, the identification of a range of autoantibodies acting on specific synaptic sites in the brain has been an important development as these disorders can present initially to psychiatrists. This requires psychiatrists to remain vigilant for the possibility of autoimmune disorders mimicking psychiatric syndromes (Davison, 2012; Rosenfeld and Dalmau, 2011). Because the brain can be affected by various autoimmune processes, it is possible that some psychiatric disorders may have an autoimmune basis. We review the literature on peripheral and central nervous system (CNS) immune dysregulation and autoimmunity in bipolar disorder. The mechanisms of the innate and adaptive immune systems in the pathophysiology of bipolar disorder are explored. The clinical features and pathogenesis of neuropsychiatric systemic lupus erythematosus, anti-NMDA encephalitis, and Hashimoto’s encephalopathy are summarized.
Evidence of immune dysregulation in bipolar disorder
The immune system can broadly be divided into the innate and adaptive immune systems, or cell-mediated immunity and humoral immunity. The innate immune system is the first and immediate line of defence against pathogens; it consists of monocytes, monocyte-derived dendritic cells and macrophages, neutrophils, natural killer cells, mast cells, and various cytokines and chemokines, such as interferon (IFN) γ, interleukin (IL) 1, IL-6, and tumour necrosis factor α (TNF-α) (Fearon and Locksley, 1996). By contrast, adaptive immunity occurs after a latent phase and is primarily activated by antigen-presenting cells, which include macrophages and dendritic cells (Nguyen et al., 2002). The adaptive immune response, once activated through antigen recognition receptors on the surface of T cells called pattern recognition receptors, is mediated by T (CD4 and CD8) and B lymphocytes. CD8 T cells are cytotoxic and are responsible for destroying intracellular pathogens, whereas CD4 T cells or T-helper cells play critical roles in immunomodulation through the secretion of cytokines which are signalling molecules involved in the on-going activation and recruitment of immune cells (Zhu and Paul, 2008). The four key CD4 T cells are Th1, Th2, Th17, and Treg cells. Th1 cells release pro-inflammatory cytokines, such as IFN γ, TNF, and IL-2; Th2 cells release IL-4, IL-5, and IL-10, which act as regulators and anti-inflammatory cytokines and are responsible for activation of humoral immunity via the activation of B cells through IL-4 (Bellone, 2005; Mosmann and Sad, 1996; Waite and Skokos, 2012). Th17 and Treg cells play important but different roles in the pathogenesis of autoimmunity (La Cava, 2009; Stockinger and Veldhoen, 2007). T cells and cytokines thus form an important component of cell-mediated immunity. An optimal balance between anti-inflammatory and pro-inflammatory cytokines is vital for the optimal functioning of the immune response (Ng et al., 2003). B cells, however, play an important role in antibody-mediated or humoral immunity through the production of antibodies against foreign antigens.
The majority of the studies on bipolar disorder have focussed on cell-mediated immunity by measuring inflammatory markers and cytokines, finding specific patterns of cytokine levels. A recent large meta-analysis showed elevated levels of TNF-α, soluble tumour necrosis factor receptor type 1 (sTNF-R1) and soluble interleukin-2 receptor (sIL-2R) in manic patients, compared to healthy control subjects and euthymic bipolar patients. The levels of sTNF-R1 were also elevated in bipolar euthymic patients compared to healthy control subjects (Munkholm et al., 2013). In addition to the presence of elevated acute-phase proteins, including haptoglobin and C-reactive protein, complement levels, such as C3C and C4 concentrations, have been found to be elevated in bipolar disorder (Maes et al., 1997; O’Brien et al., 2006; Wadee et al., 2002). There appears to be a phasic difference in the level of cytokines in manic bipolar patients, who show elevated plasma levels of TNF-α, IL-2, and IL-4 (Ortiz-Dominguez et al., 2007). Increases in the levels of IL-8 and IL-6 have been reported in depressed bipolar patients (Brietzke et al., 2009). There is evidence of stage-related abnormalities in cytokines; all interleukins and TNF-α are elevated in the early stages of illness, whereas in the later stages of illness, TNF-α and IL-6 continue to be elevated, but IL-10 does not. Thus, these cytokines may serve as important markers of disease progression in bipolar disorder (Kauer-Sant’Anna et al., 2009). Berk et al. (2011) have hypothesized that inflammatory mediators are involved in the episode-related cognitive decline seen in bipolar disorder. Bipolar patients in the euthymic state also have abnormalities in their chemokine levels that induce chemotaxis (i.e. the movement of leukocytes towards sites of inflammation), indicating ongoing inflammation in a clinically quiescent stage of illness (Brietzke et al., 2009). Leptin is an adipocyte-derived cytokine that is of interest in the pathogenesis of bipolar disorder as it plays an important role in obesity and promotes Th1 cell differentiation modulating immune responses (La Cava and Matarese, 2004). Insulin-like growth factor (IGF)-1 has been implicated in the pathogenesis of bipolar disorder, insulin resistance, and immune dysfunction and may be a future biomarker of interest (Smith, 2010; Squassina et al., 2013).
Does neuroinflammation play a role in the pathophysiology of bipolar disorder?
Neuroinflammation is the result of an immune response in the CNS that involves a complex interaction between the CNS innate immune system, the blood–brain barrier (BBB) and the peripheral immune system. Microglia are the first line of defence in the CNS. Neuronal injury, infection, or ischaemia can activate microglia through specialized receptors on their surface called pattern recognition receptors. Three major families of pattern recognition receptors exist; Toll-like receptors (TLR), Nod-like receptors (NLR), and RIG1 like receptors, which recognize specific exogenous and endogenous ligands activating specific inflammatory pathways (Lampron et al., 2013). TLR and NLR can recognize pathogen-associated molecular patterns (PAMPs) during infection or damage-associated molecular patterns (DAMPs) such as intracellular protein and non-protein products and nucleic acids during neuronal injury. The engagement of microglial TLR activates transcription factors such as NF-KB and mitogen-associated protein kinase leading to the release of inflammatory cytokines (e.g. IL-1β, TNF-α, IL-6), chemokines acting as chemoattractants for other immune cells, reactive oxygen species, nitric oxide, and prostanoids by activating the phospholipase A2 and COX-2 pathway. Although microglial activation is an important component of innate immunity serving to protect the CNS from harmful stimuli, under some poorly understood circumstances, microglia can become overactivated and cause neurotoxicity and neurodegeneration. Two postulated signals are direct stimulation by endogenous proteins or environmental toxins and neuronal damage-released DAMPs recognizable by microglial TLR (Block et al., 2007; Ransohoff and Brown, 2012).
Activated microglia express indoleamine 2,3-dioxygenase, which diverts the tryptophan pathway towards producing tryptophan catabolites: of these, 3-HK generates free-radical species that can cause oxidative stress and lipid peroxidation, whereas quinolic acid is associated with glutamate-induced oxidative damage via NMDA receptor-mediated, calcium-dependent uncoupling of neuronal mitochondrial electron transport (Berk et al., 2011; Dugan et al., 1995; Najjar et al., 2013). The cytokine IL-1β increases NMDA receptor (NMDAR) contributing to glutamate-mediated neurotoxicity (Viviani et al., 2003). Kaindl et al. (2012) showed that activation of the NMDAR on microglia triggers inflammation and cell death, which in turn can further activate microglia and initiate a forward feedback loop (Kaindl et al., 2012). Glutamate can activate a Th1 response, and it plays an important role in immunomodulation, particularly the initiation and development of T cell-mediated immunity in the periphery and CNS (Pacheco et al., 2007). Cytokines can upregulate neuronal serotonin transporter activity reducing the strength of serotonin activity at serotonin receptors (Mössner et al., 1998; Zhu et al., 2005). Cytokines such as IL-6 and TNF-α activate the hypothalamic–pituitary–adrenal axis, increasing cortisol levels which can be neurotoxic and may be central to the pathogenesis of depressive symptoms and cognitive deficits (Daban et al., 2005; Dunn, 2000). Elevated cortisol levels reduce postsynaptic serotonin receptors, serotonin responsiveness, and brain-derived neurotrophic factor levels, thus affecting neurotransmitter regulation and neuroplasticity (Huang and Reichardt, 2001; Issa et al., 2010; Miller et al., 1999). The strongest clinical evidence for the pathogenic role of cytokines comes from the effects of IFN-α in the treatment of hepatitis C and cancer (Renault et al., 1987). IFN-α administration is associated with widespread bilateral increases in glucose metabolism in subcortical regions including the basal ganglia (Capuron et al., 2007) and anterior cingulated, which are associated with fatigue and cognitive effects respectively (Capuron et al., 2005).
Microglia interact closely with other glial cells, namely astrocytes and oligodendrocytes, that play important roles in neural, cognitive, and behavioural functions (Najjar et al., 2013). Astrocytes maintain BBB integrity, regulate synaptic transmission, and are responsible for downregulating harmful inflammation initiated by microglia by activating the Th2 response and D2 receptors. Astrocytes express TLR and can amplify the CNS innate immune response by secreting complement components and chemokines. Astrocytes reduce the effects of excitotoxic glutamate by increasing their uptake, and inflammatory responses in the CNS can compromise this essential astrocyte function, thus amplifying excitotoxicity in the CNS (Zuo et al., 2010; Luo and Chen, 2012). Cross-talk between microglia and astrocytes has been implicated in several neurological conditions, but the details of this interaction are still unclear (Liu et al., 2011b).
The third glial cell, the oligodendrocyte, is actively involved in the process of myelination and can downregulate the inflammatory response of microglia by secreting anti-inflammatory cytokines (Edgar and Sibille, 2012; Najjar et al., 2013). TNF-α has a direct toxic effect on oligodendroglia, potentially contributing to apoptosis and demyelination (Miller et al., 2009), and it is inversely related to the brain-derived neurotrophic factor concentration, which is known to be reduced in bipolar disorder (Brietzke and Kapczinski, 2008; Kapczinski et al., 2009; Lin, 2009). Oligodendrocytes are particularly susceptible to oxidative stress and mitochondrial injury as they contain low levels of antioxidant glutathione and express NMDAR which exposes them to glutamate excitotoxicity (Bradl and Lassmann, 2010). The release of reactive oxygen species and nitric oxide not only amplifies the release of pro-inflammatory cytokines, but reactive oxygen species also leads to mitochondrial injury by activating oxidative and apoptotic pathways (Lopez-Armada et al., 2013) creating a vicious cycle of inflammation, oxidative and nitrosative stress (O&NS), and glutamate excitotoxicity. Furthermore, the activation of the O&NS pathways can damage membrane fatty acids and anchorage molecules such as palmitic acid and myristic acid, causing aberrations in cell signal transduction, cellular architecture, cell differentiation, and neuroprogression (Maes et al., 2012a).
The innate immune response can be further amplified by a breakdown in the BBB. Leukocytes in the peripheral circulation can bind to the endothelial cells of the BBB through adhesion molecules such as VCAM-1 and ICAM-1 and can pass through the BBB due to a loosening of the endothelial tight junctions (Rossi et al., 2011). Peripheral cytokines can enter the BBB through areas of the brain that lack the BBB, such as the circumventricular organs (Dantzer et al., 2000), and the vagus nerve acts as a connection between the peripheral immune system and the brain (Gidron et al., 2007). In the CNS, inflammation results in the production of inflammatory mediators such as TNF-α, IL-6, IL-1β, and arachidonic acid (AA), which can increase BBB permeability (Shalev et al., 2009). Locally produced cytokines can also either activate neurons that project to specific brain areas or diffuse into the brain parenchyma by volume transmission to reach their targets (Dantzer et al., 2000). In summary, microglial activation results in the activation of an inflammatory cascade that produces a range of downstream mediators that can have the net effect of causing oligodendrocyte apoptosis, BBB damage, mitochondrial dysfunction, and neuronal injury.
Studies have shown evidence of neuroinflammation in individuals with bipolar disorder. Postmortem studies in bipolar disorder have shown the presence of excitotoxic markers and neuroinflammation in the frontal cortex with activation of the IL-1 receptor cascade (Rao et al., 2010). The cytokine IL-1β, which is indicative of microglial activation, was found to be elevated in the CSF of euthymic patients who had a recent manic or hypomanic episode (Soderlund et al., 2011). The cingulate cortex shows evidence of upregulation of the initiating step of the kynurenine pathway and increased cell adhesion molecule immunoreactivity, indicating neuroinflammation (Miller et al., 2006; Thomas et al., 2004). Zanetti et al. (2009), using diffusion-tensor imaging, found disruptions in the ventromedial prefronto-limbic-striatal white matter in bipolar depression, which were hypothesized to result from inflammatory causes (Zanetti et al., 2009). Bipolar-type presentations with mania and hypomania have been described with IFN-α (Chang et al., 2005; Constant et al., 2005; Iancu et al., 1997; Malek-Ahmadi, 2001). Elevated levels of glutamate, markers of oxidative stress, and mitochondrial dysfunction are found in the brains of individuals with bipolar disorder (Andreazza et al., 2008; Clay et al., 2011; Gigante et al., 2012; Hashimoto et al., 2007).
Glial cell abnormalities have been found; however, the data are not consistent and are affected by confounders such as medication and phases of illness (Najjar et al., 2013). Astrocyte involvement is indicated by decreased glial fibrillary acidic protein mRNA in the cingulate cortex (Webster et al., 2005). Reduced oligodendrocyte density, decreased oligodendrocyte gene expression in the frontal cortex, and white matter involvement have also been detected. (Edgar and Sibille, 2012). White matter hyperintensities, which can indicate abnormal myelination, are found early on in bipolar disorder and play an important role in the pathophysiology of bipolar disorder through a postulated role of disconnectivity between the frontal cortex and regions controlling emotional regulation (McIntosh et al., 2008; Mahon et al., 2010; Manji et al., 2000). Further evidence for glial cell loss comes from the levels of S100B, which is actively released by astro- and oligodendrocytes. Significant increases in serum S100B have been observed during episodes of mania and depression, although not in euthymic patients; S100B has therefore been proposed as a biomarker for mood disorders (Schroeter et al., 2013). Individuals with bipolar disorder show a marked increase in the upregulation of pro-apoptotic genes along with a downregulation of antioxidant genes, inducing vulnerability to apoptosis and oxidative stress-induced damage (Benes et al., 2006). The combination of neuronal damage through the above mechanisms and the suppression of neurogenesis (Das and Basu, 2007; Losif et al., 2006) creates an environment biased towards neuroprogression that may be implicated in the pathogenesis of symptoms of bipolar disorder. A detailed explanation of the interaction between cytokines, O&NS stress, mitochondrial dysfunction, apoptotic pathways, and neurogenesis is outside the scope of this review and we refer readers to the articles by Berk et al. (2011) and Maes et al. (2011) for further reading.
Interestingly, the above-described pathways, namely mitochondrial dysfunction, neuroinflammation, and oligodendrocyte abnormalities, show parallels with multiple sclerosis (Konradi et al., 2012), which makes it important to examine the role of autoreactive T and B cells in the pathophysiology of bipolar disorder (discussed in the next section). Because cytokines play a crucial role in the development, differentiation, and regulation of immune cells, the dysregulation of cytokine production is thought to have an important role in the development of autoimmune disease (O’Shea et al., 2002). TLR and NLR are an important link between innate and adaptive immunity and may be responsible for the initiation of the autoimmune response (discussed later). Moreover O&NS and apoptotic pathways release immunogenic self-epitopes from damaged membranes and proteins that can result in secondary autoimmune reactions (Maes et al., 2011).
Peripheral autoimmunity and bipolar disorder
Autoimmunity is defined as a process whereby an immunological response is directed against the body’s self-antigens. Autoimmune disease occurs when the immune reaction overwhelms the normal tolerance checkpoints and occurs even in the absence of an offending antigen (Fairweather, 2007; Goodnow, 2007). A common feature of autoimmune diseases is the presence of autoantibodies, which are important markers in the diagnosis and classification of autoimmune diseases and can be useful in psychiatric clinical practice to detect an autoimmune process. Autoimmune tissue damage can be cell-mediated via T cell activity (cell-mediated immunity), antibody-mediated via B cells (humoral immunity), or both (Bellone, 2005; Fairweather, 2007). TLR on the surface of T and B cells are considered to play an important role in the initiation and progression of systemic autoimmune disease through recognition of exogenous and endogenous antigens (Marshak-Rothstein, 2006). DAMPs released during mitochondrial apoptosis such as oxidized mitochondrial DNA bind to NLR activating the inflammatory response through caspase-1 activation (Lopez-Armada et al., 2013). T cell-mediated damage to tissues occurs through the release of cytotoxic cytokines, prostaglandins, and reactive nitrogen and oxygen species. Individuals with bipolar disorder have a bias towards a Th1 pro-inflammatory profile and a reduced number of Treg cells, along with higher levels of senescence-associated cells such as CD8 and CD25 T cells (do Prado et al., 2012). The T cell system in bipolar disorder is activated in both symptomatic and euthymic patients with bipolar disorder (Breunis et al., 2003). Specific alterations in TLR agonist-mediated activity were found in bipolar patients (McKernan et al., 2011). Infections are known to activate TLRs, and antibodies to toxoplasma and cytomegalovirus have been found in bipolar disorder patients (Tedla et al., 2011). Following TLR-mediated antigen recognition, GSK3 plays a crucial role in the immune response, and lithium is a GSK3 inhibitor capable of reducing inflammation (Beurel et al., 2010). The monocytes of a large proportion of bipolar patients and the offspring of bipolar parents show an inflammatory gene expression signature (Padmos et al., 2008) and are believed to arise from a shared environmental factor (Padmos et al., 2009). In addition to the enhancement of pro-inflammatory monocytes, Tregs are also enhanced in bipolar disorder, except in individuals with autoimmune thyroid disease or bipolar disorder who demonstrate a significantly reduced percentage of Tregs (Drexhage et al., 2011). In summary, abnormalities in T cells and cytokines involved in the Th1/Th2 balance show an imbalance (Brietzke et al., 2011; Kim et al., 2007) and may represent underlying T and B cell autoimmunity.
B cell-mediated immunity, however, occurs primarily through antibodies. IL-4 released by Th2 cells activates B cells to produce antibodies. In some individuals who are susceptible due to a breakdown in tolerance, B cells may produce self-reactive autoantibodies, which constitute the hallmark of autoimmune disease. Self-reactive antibodies can induce tissue damage by binding to self-tissues and activating complement-mediated damage, facilitating antibody-dependent cell-mediated cytotoxicity (ADCC), forming immune complexes, and initiating the complement-induced lysis or removal of cells by phagocytic immune cells (Fairweather, 2007).
Thyroid autoimmunity has particularly been shown to be an independent risk factor for bipolar disorder with no association with lithium exposure (Haggerty Jr et al., 1990, 1997; Kupka et al., 2002), and it may serve as an endophenotype for bipolar disorder (Vonk et al., 2007). Bipolar disorder is associated with organ-specific autoimmunity to the antigens TPO, H/K ATPase, and GAD65, and the level of TPOAb is inversely correlated with the level of T cell activation (Padmos et al., 2004). The female offspring of bipolar disorder patients are more vulnerable to developing thyroid autoimmunity, as evidenced by elevated TPOAb titres. However, TPOAb-positive offspring do not show an increased prevalence of mood disorders compared to TPOAb-negative offspring (Hillegers et al., 2007). Sidhom et al. (2012) have shown that patients with bipolar disorders have significantly more autoantibodies compared to people with other affective or psychotic disorders and non-psychiatric controls. They concluded that a possible autoimmune activation occurs in at least a subgroup of psychiatric patients, particularly among those suffering from bipolar disorder. Spivak et al. (1996) found an increased frequency of antinuclear antibody (ANA) positivity in bipolar patients in comparison to controls, independent of any lithium therapy. Considering the above evidence, the association is strongest for thyroid autoimmunity, but its pathophysiology is uncertain and may be related to cross-reactivity with glutamate receptors (see later).
The role of CNS autoimmunity in bipolar disorder
The cells of the CNS innate immune system interact with T cells in the periphery to activate the adaptive immune system. Dendritic cells in the perivascular space and meninges can activate T cells in the periphery that then traverse the BBB. Once in the CNS, CD4 T cells can be restimulated by perivascular macrophages and dendritic cells, which act as antigen-presenting cells. T cells release Th1- and or Th2-type cytokines upon antigen recognition by TLRs and interact with microglia and astrocytes to amplify the immune response, resulting in the clearance of the offending antigen (Aloisi et al., 2000).
The inflamed brain provides a conducive environment for B cell survival and proliferation resulting in the production of antibodies (Corcione et al., 2005). B cells are known to cross the CNS and can initiate an autoimmune response against endogenous and exogenous ligands through mechanisms mentioned earlier (Meinl et al., 2006). The production of autoreactive antibodies against neuronal antigens (brain reactive antibodies) can interfere with neuronal transmission, prevent binding of neurotrophic factors such as brain-derived neurotrophic factor, and cause direct neuronal injury (Kapadia and Sakic, 2011). For example, autoantibodies directed against an epitope on the extracellular surface of oligodendrocytes can induce demyelination either through activation of complement or through their recognition by Fc-receptors of activated macrophages (Bradl and Lassmann, 2010). T and B cell autoimmunity can be triggered through several mechanisms. PAMPs can cross-react with self-epitopes through a process of molecular mimicry (Chastain and Miller, 2012). Secondly, cell injury in the CNS can result in the release of DAMPs that can become immunogenic, initiating a T cell autoimmune response against self-antigens (Harris and Fabry, 2012). Extracellular mitochondrial DNA is known to be extremely immunogenic and can directly initiate an autoimmune response (Zhang et al., 2010). Cross-reactivity with self-antigens can initiate an autoimmune reaction against neuronal antigens. For example, a subset of the anti-DNA antibody is known to cross-react with the NR2 glutamate receptor (DeGiorgio et al., 2001); elevated serum antibodies to the NR2 subunit of the glutamate receptor are found in the acute phase of mania (Dickerson et al., 2012). Some DAMPs, such as S100B, extracellular nucleotides, and extracellular matrix components of basement membrane are plausible candidates in the initiation of autoimmune response. Furthermore, microscopic particles have been found in the CSF of individuals with bipolar disorder and only rarely in controls. It is unclear whether these particles represent DAMPs, and further research is required to clarify this (Johansson et al., 2012). Kapadia and Sakic (2011) have integrated the CNS innate and adaptive immune system mechanisms by proposing two phases, the inflammatory phase, which is dominated by innate immunity and leads to functional damage, followed by the autoimmune phase, which is responsible for structural damage such as neurodegeneration and atrophy. CNS T and B cell autoimmunity is an underexplored area in bipolar disorder. It should be noted that endogenous protective regulatory signals, such as Treg cells and B cell-mediated immunoregulation, exist in the brain to prevent autoimmune responses and that the dysregulation of such mechanisms may be responsible for initiating and promoting autoimmune responses (Block et al., 2007; Goverman, 2009).
Autoimmune disorders and bipolar disorder: focus on neuropsychiatric lupus erythematosus, anti-NMDA encephalitis, and Hashimoto’s encephalopathy
A large Danish cohort study showed that autoimmune processes precede the onset of bipolar disorder (Eaton et al., 2010). Although research on the link between autoimmune diseases and psychiatric disorders is at a relatively early stage, autoimmune diseases, such as autoimmune synaptic encephalitis, neuropsychiatric systemic lupus erythematosus (NPSLE), and encephalopathy associated with autoimmune thyroiditis (EAAT), particularly Hashimoto’s encephalopathy, have been studied in greater depth by rheumatologists and neurologists. Although case reports of other autoimmune diseases presenting with symptoms of bipolar disorder exist, we focus on these three disorders due to their high biological plausibility of being related to the pathophysiology of bipolar symptoms based on evidence of involvement of the glutamate pathway. Thus, these should be considered high on the differential diagnosis list when autoimmunity is suspected in psychiatry. The three autoimmune disorders are summarized in Table 1.
Summary of important autoimmune disorders presenting with psychiatric symptoms.
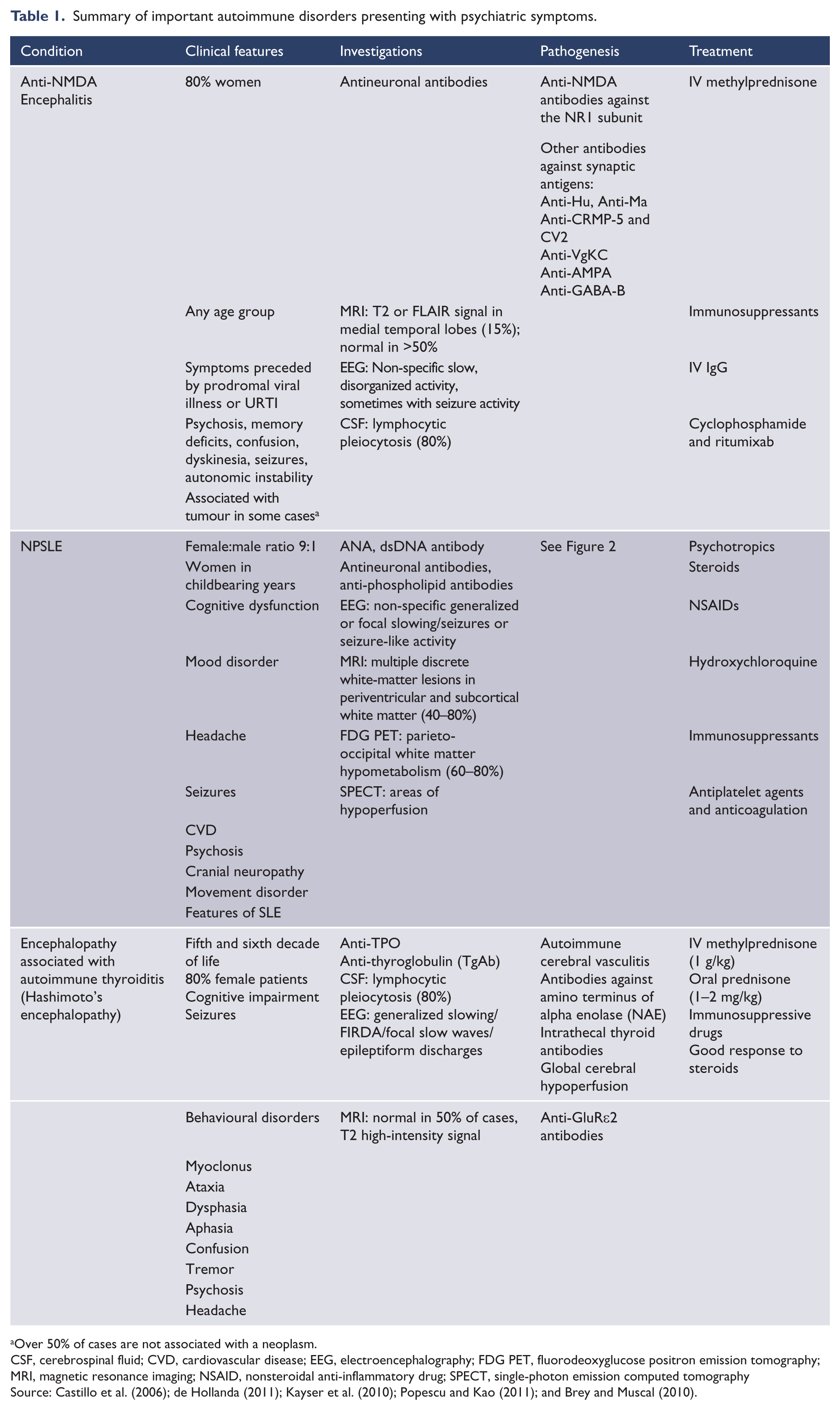
Over 50% of cases are not associated with a neoplasm.
CSF, cerebrospinal fluid; CVD, cardiovascular disease; EEG, electroencephalography; FDG PET, fluorodeoxyglucose positron emission tomography; MRI, magnetic resonance imaging; NSAID, nonsteroidal anti-inflammatory drug; SPECT, single-photon emission computed tomography
Source: Castillo et al. (2006); de Hollanda (2011); Kayser et al. (2010); Popescu and Kao (2011); and Brey and Muscal (2010).
Among the best-known and most well-studied conditions is NPSLE. The prevalence of neuropsychiatric manifestations in SLE is estimated to range from 14% to 80% in adults and from 22% to 95% in children (Muscal and Brey, 2010; Popescu and Kao, 2011). There have been case reports of NPSLE presenting as bipolar disorder (Alao et al., 2009; Yeh and Hu, 2007). The most common symptoms in NPSLE include the following: cognitive dysfunction (75–80%), mood disorders (69–74%), headache (39–61%), seizures (8–18%), cerebrovascular disease (2–8%), psychosis (3–5%), cranial neuropathy (1.5–2.1%), and movement disorders (1%). These symptoms indicate that NPSLE may present to psychiatrists first (Popescu and Kao, 2011). Cytokines and chemokines play key roles in the pathogenesis of NPSLE (Okamoto et al., 2010). A key effector mechanism in NPSLE in the development of cognitive dysfunction and mood disorders may be the glutamatergic pathway through the NMDA receptors situated in the hippocampus, frontal regions, and limbic system (Mak et al., 2009). dsDNA antibody is an important antibody in this respect due to its cross-reactivity with the NR2 glutamate receptor (DeGiorgio et al., 2001). The pathogenesis of NPSLE is multifactorial and is shown in Figure 1.
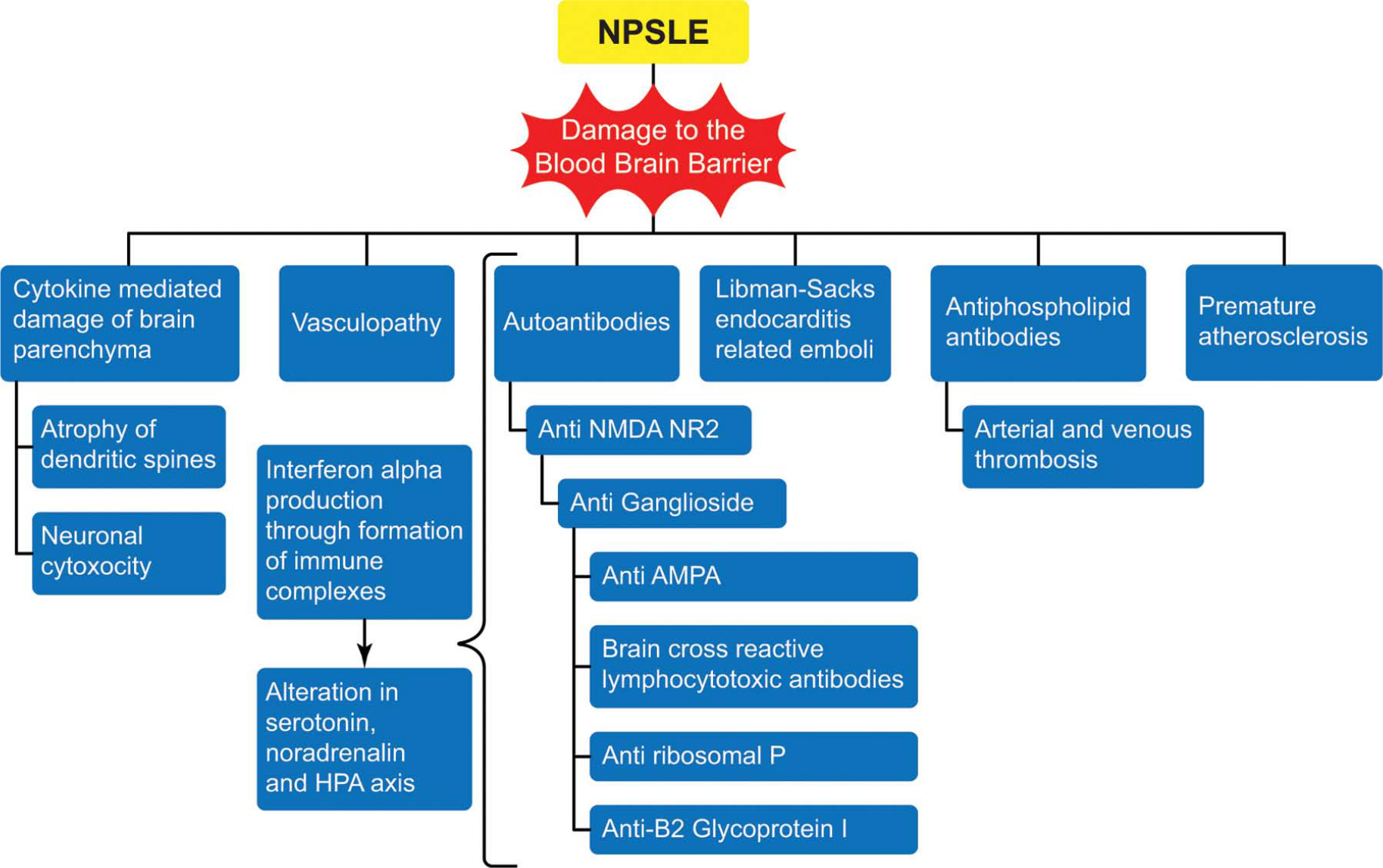
The pathogenic mechanism of autoantibodies is related to the breakdown of the blood–brain barrier and CSF autoantibodies forming and binding to the cell antigens that are released by neurocytotoxic antibodies (e.g. anti-ribosomal P, anti-NR2), which form immune complexes and stimulate production of IFN-α and pro-inflammatory cytokines and chemokines (Fragoso-Loyo et al., 2007; Santer et al., 2009). IFN-α is thought to be the effector mechanism of cognitive and mood effects in NPSLE. Thus, serum antibodies of SLE (antinuclear antibody and dsDNA antibody) may not be reliable markers of neuropsychiatric disease, as there is a lack of correlation between serum autoantibodies and NPSLE manifestations, and neuropsychiatric manifestations may be mediated by alternate antibodies, such as NR2 glutamate-receptor antibodies that cross-react with anti-DNA antibodies (Arinuma et al., 2008; DeGiorgio et al., 2001; Omdal et al., 2005).
The diagnosis of neuropsychiatric lupus in clinical practice is a contentious issue amongst psychiatrists, rheumatologists, and other caregivers. Standard laboratory testing and imaging studies may not always detect active brain disease or static damage (Wright, 2010). Thus, it is important for psychiatrists to be aware of the limitations in diagnostic criteria and the potential management issues they face. There is no single diagnostic test for SLE, and the American College of Rheumatology criteria are intended for classification rather than diagnosis (Hughes, 1998). ANA and dsDNA antibody are sensitive and specific serological markers, respectively, for SLE. The application of the NPSLE criteria in psychiatry is limited by the requirement of a temporal relationship between SLE and a psychiatric disorder (SLE before neuropsychiatric presentation); this contradicts evidence showing that the brain may be affected early in SLE with primary neuropsychiatric presentations, even before a diagnosis of SLE is made, and neither a normal erythrocyte sedimentation rate nor negative serology excludes CNS lupus (Joseph et al., 2007; Petri et al., 2008). Moreover, NPSLE manifestations have been found to be negatively correlated with systemic manifestations such as joint and skin involvement in children and adults (Harel et al., 2006; Karassa et al., 2000), and the American College of Rheumatology criteria may be biased towards more severe disease and a longer duration of disease and do not reflect clinical diagnoses as assigned by lupus experts (Wallace and Hahn, 2006). Thus, clinical diagnostic impressions in NPSLE must be based on the combined use of immunoserological testing, functional and/or structural neuroimaging, and standardized neurological, rheumatological, psychiatric, and neuropsychological assessments (Hermosillo-Romo and Brey, 2002). These findings show that the current criteria for NPSLE diagnosis and their generalizability to psychiatric patients should be examined in view of the high degree of morbidity and mortality associated with NPSLE (Monov and Monova, 2008). The major histocompatability complex located on chromosome 6 provides further support for the association between bipolar disorder and NPSLE: the genes in this region have been implicated in bipolar disorder and provide a strong genetic association with a range of autoimmune disorders, particularly SLE (Harley, 2002; International Schizophrenia Consortium et al., 2009; Jonsen et al., 2004).
The identification of encephalitis associated with antibodies against cell surface and synaptic antigens has had a substantial impact in neuropsychiatry. Classically, encephalitis due to synaptic antibodies evolves over days to weeks and includes psychiatric manifestations as diverse as irritability, mood disturbance, hallucinations, and personality disturbances. Further neurocognitive changes occur in the form of short-term memory loss, sleep disturbances, and seizures. Anti-NMDA encephalitis is of particular relevance to psychiatrists due to its clinical presentation. Over 200 patients with anti-NMDA encephalitis have been described, and 75% present initially to psychiatrists (Kayser et al., 2010). The antibody in anti-NMDA encephalitis specifically recognizes the NR1 subunit of the NMDA receptor, which is present in all NMDA receptors (Dalmau et al., 2008). In contrast, the antineuronal antibodies in NPSLE recognize the NR2A and NR2B subunits of the NMDA receptor, which are highly concentrated in the hippocampus (Kayser and Dalmau, 2011). These differences in location and antigenic targets may explain the difference in presentation between NPSLE and anti-NMDA encephalitis. Recently, antineuronal antibodies, which are characteristic of synaptic encephalitis, have been considered important biomarkers in NPSLE (Gorman, 2011).
EAAT is the third important differential. In the context of encephalopathy, anti-TPO antibodies are used to diagnose Hashimoto’s encephalopathy (Mocellin et al., 2007). Reports exist of Hashimoto’s encephalopathy presenting with depression, mania, and cognitive dysfunction (Bochetta et al., 2007; Canelo-Aybar et al., 2011; Liu et al., 2011a; Mussig et al., 2005). This disorder is also termed steroid-responsive EAAT due to its often-dramatic response to steroids (Castillo et al., 2006). Despite concerns regarding steroid use in psychosis, a good response to steroids has been obtained in psychosis associated with autoimmune thyroiditis, implying an immunological pathogenesis (Arrojo et al., 2007; Gómez-Bernal et al., 2007). A recent study showed that anti-GluRϵ2 antibodies may be associated with the neuropsychiatric manifestations of psychiatric patients with antithyroid antibodies (Chiba et al., 2012). The treatment response to immunomodulation in these patients has not been studied.
In Table 2, we present a list of clinical features based on a review of the important autoimmune conditions described above that can help increase the detection of autoimmunity in psychiatry. Although this list has not yet been validated to any degree, it may help clinicians to suspect autoimmunity and form a basis for further research. The detailed treatment of each disorder is outside the scope of the article and includes a combination of psychotropics, corticosteroids, and immunosuppressants that is summarized in Table 1.
Clinical indicators that should raise suspicion of autoimmunity in practice.
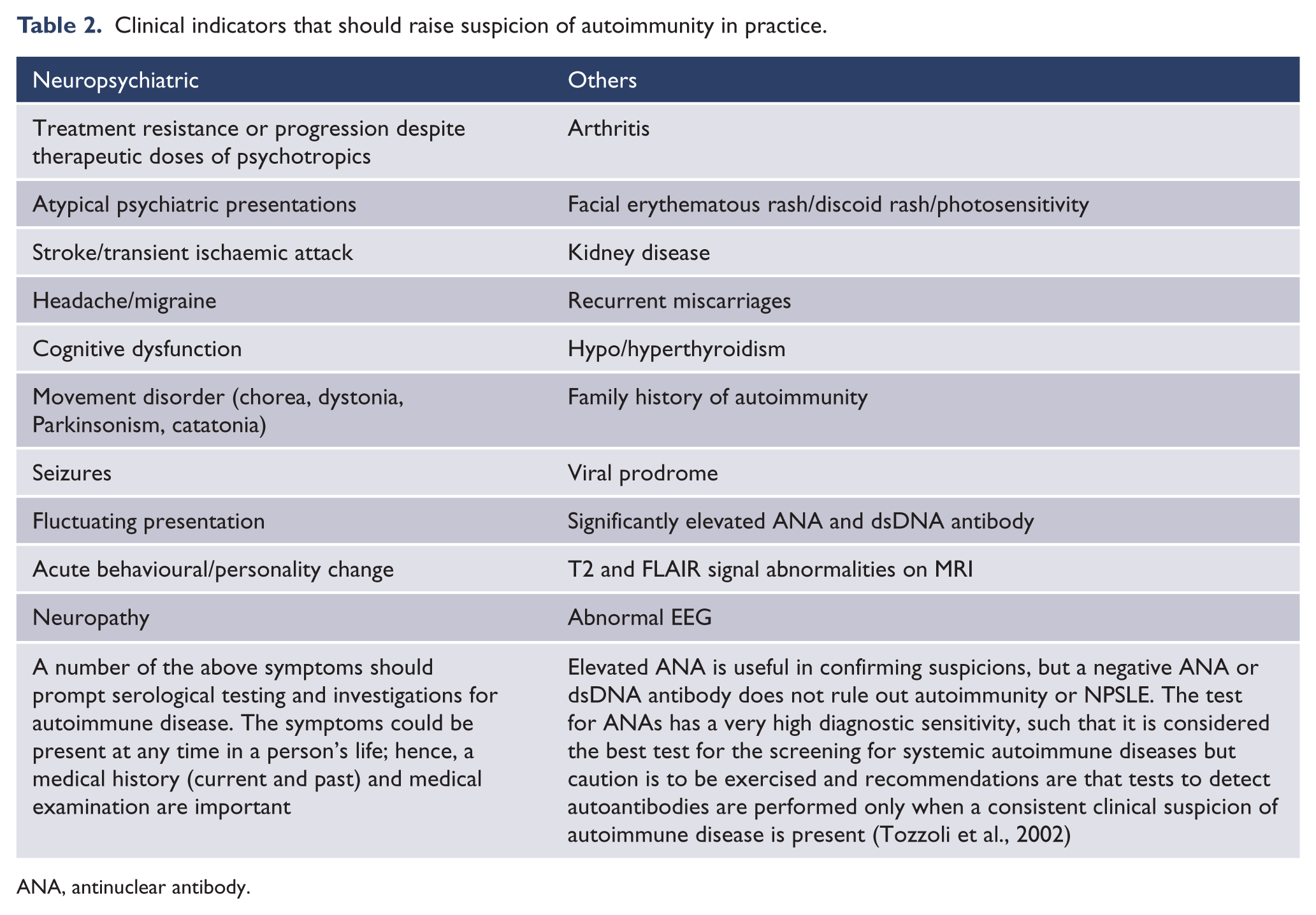
ANA, antinuclear antibody.
Thus, NPSLE, some forms of autoimmune encephalitides, and some subsets of bipolar disorder presentations may have common underlying pathophysiological mechanisms.
The role of stress and its interaction with the immune system
A stressful event is often associated with bipolar disorder presentations. There are no specific studies examining the role of stress, immune dysregulation, and autoimmunity in bipolar disorder, and we discuss this association in general terms. The link between stress and mood disorders is well recognized, with corticotrophin-releasing hormone playing an important role in the pathophysiology (Stout and Nemeroff, 1994). Retrospective studies in autoimmune diseases have found that up to 80% of patients report uncommon emotional stress before disease onset (Stojanovich and Marisavljevich, 2008). Psychosocial stressors activate corticotrophin-releasing hormone and the sympathetic nervous system, increasing NF-KB DNA binding in inflammatory cells and resulting in the release of cytokines that act through the pathways described earlier. Pro-inflammatory cytokines then induce cortisol release, which aims to decrease the inflammatory response. This pathway is downregulated in chronic stress, resulting in an imbalance between pro-inflammatory and anti-inflammatory cytokines (Miller et al., 2009). Furthermore, there is evidence that brief naturalistic stressors, such as academic exams, potentiate a shift towards a Th2 response, whereas chronic stress reduces both Th1 and Th2 responses (Plotnikoff, 2007; Segerstrom and Miller, 2004). Figure 2 synthesizes this evidence to propose a hypothesis of the interaction between stress/trauma, cytokines, the hypothalamic–pituitary–adrenal axis, autoimmunity, and the brain in the pathogenesis of psychiatric symptoms. The model is complicated by other triggers, such as physical CNS injury and infections, both of which can activate autoimmunity. This model can be extrapolated to other psychiatric disorders. We refer readers to Plotnikoff (2007) and Segerstrom and Miller (2004) for a more detailed explanation on the role of stress and the immune system.
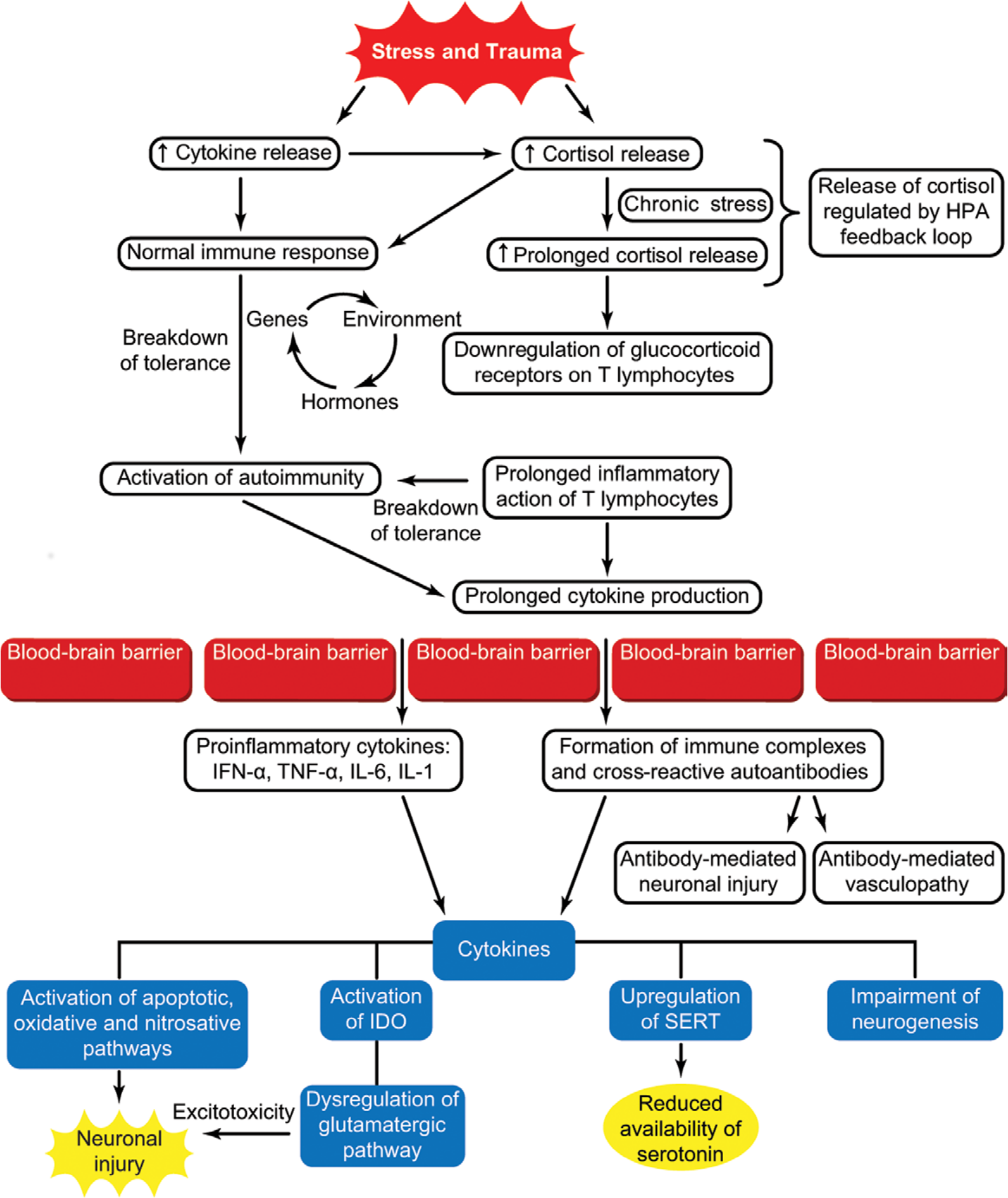
The role of stress and its interaction with the immune system. Stress and trauma lead to activation of cytokine and cortisol release which lead to an adaptive T cell response to external or internal antigens (Segerstrom and Miller, 2004). Cortisol acts by reducing inflammatory tissue damage due to inflammatory cytokines. The cortisol release is modulated by the hypothalamic–pituitary–adrenal axis feedback mechanism to avoid prolonged cortisol release. In chronic stress, however, prolonged elevated cortisol release leads to downregulation of cortisol receptors on T lymphocytes resulting in unrestricted tissue damage and amplification of the immune response resulting in autoimmunity in susceptible individuals (Cohen et al., 2012). This susceptibility is governed by genes, environmental factors, or hormonal factors. The activation of autoimmunity and cytokine release can result in endothelial cell damage at the level of the blood–brain barrier resulting in passage of cytokines through the blood–brain barrier. Cytokines then act on the brain though various mechanisms resulting in the genesis of psychiatric symptomatology. SERT, serotonin transporter; IDO, indoleamine 2,3-dioxygenase.
Future research and implications for clinical practice
A limitation in the existing literature examining cytokines and bipolar disorder is the cross-sectional nature of the studies. It is unclear if the cytokine dysfunction represents a true causal mechanism in the pathophysiology of bipolar disorder, is an epiphenomenona, or is related to medication (Langan and McDonald, 2009). It may also represent underlying T and B cell autoimmunity. It is also too early to comment on the relationship of autoimmunity with the pathophysiology of bipolar disorder. Identifying specific autoantibodies in varying phases of the illness may be one way forward (antibodies to 5-HT and anchorage molecules as found in depression; Maes et al., 2011, 2012b). Antibodies against NMDARs merit further investigation in bipolar disorder. Future research should include autoantibodies and inflammatory markers as biomarkers in addition to clinical features and neuroimaging with the aim of stratifying psychiatric presentations. This is in line with proposals by Kapur et al. (2012) for “stratified psychiatry”, which will improve clinical outcomes across conventional diagnostic boundaries. With the advent of neuroimaging techniques such as two-/multiphoton and fluorescence microscopy, existing nuclear medicine techniques with radiolabelled ligands for cells involved in neuroinflammation, and techniques to visualize T cell movement and migration, further advances will result in an improved understanding of the role of neuroinflammation in psychiatric disorders.
Current research examining the link between autoimmune diseases and psychiatry may be susceptible to referral and spectrum bias. Longitudinal prospective cohort studies are required to establish the pathogenesis of the neuropsychiatric autoimmune disease process and evaluate the interventions that are appropriate. There is a case for multispecialty collaboration in research projects due to the multisystem involvement of autoimmune diseases. In clinical practice, the diagnosis of autoimmune encephalopathy requires a high index of clinical suspicion, as tests including serology, EEG, CSF, and MRI can be normal and do not exclude an autoimmune pathology. Psychiatrists should pay particular attention to the milder incomplete cases of anti-NMDA encephalitis (formes frustes), which may continue to be treated as psychiatric disorders (Dalmau et al., 2011). Clues to autoimmune encephalopathies often lie in the longitudinal history. Movement disorders, cognitive dysfunction, and fluctuating cognition are common in all three autoimmune encephalopathies and can be attributed incorrectly to antipsychotic medication, thus creating a protopathic bias. A detailed medical evaluation (history and examination), cognitive examination, case-note review, and corroborative history taking from the family are important in psychiatric evaluations, particularly in atypical presentations characterized by a non-response or poor response to psychotropics. Based on advice by Neuwelt and Young (2009), the management of autoimmune encephalopathies requires a team approach, including a rheumatologist, neurologist, neuroimmunologist, psychiatrist, neuropsychologist, oncologist, and primary-care physician, to formulate a proper diagnosis and treatment plan for the management of NPSLE.
From a treatment perspective, the consideration of the immune hypothesis opens up a new avenue for research into the role of immunomodulators in psychiatry. Although this review cannot make conclusive treatment recommendations, there is a case for an empirical trial of steroids in suspected cases of immune-related psychiatric encephalopathy after excluding infections, as in cases of limbic encephalitis and steroid-responsive EAAT (Bataller et al., 2007; Castillo et al., 2006). It is important that the psychiatrist be familiar with steroid administration and its side effects. Knight et al. (2007) have noted that concerns about corticosteroids in psychiatry are not well founded and are based primarily on anecdotal evidence. The prescription of psychotropics in the context of organic brain disease can increase morbidity, mortality, and the risk of Neuroleptic Malignant Syndrome (NMS) (Adnet et al., 2000; Maat et al., 2012), notwithstanding the morbidity and mortality from untreated conditions such as NPSLE (Yu et al., 2006) and autoimmune synaptic encephalitis. Thus, a careful, individualized risk–benefit analysis is required. Among psychotropics, lithium and clozapine (used in certain treatment-resistant illnesses) have immunosuppressive properties (Beurel et al., 2010; Boufidou et al., 2004; Knight et al., 2007; Maes et al., 2007) that possibly contribute to their superiority in treatment-resistant cases; however, their use in autoimmune encephalopathies has not been evaluated. The use of anti-inflammatory medications is also known to restore adult hippocampal neurogenesis and thus has the potential to improve cognitive deficits (Monje et al., 2003). Existing cytokine inhibitors (anti-TNF-α), such as etanercept, adalimumab, and infliximab, are used in the management of rheumatoid arthritis and psoriasis and have been shown to both relieve the symptoms of psoriasis and improve affective, somatic, and cognitive functions (Soczynska et al., 2009; Tyring et al., 2006), but they have not been evaluated in primary psychiatric presentations. The role of smoking cessation and addressing alcohol misuse should not be overlooked, as smoking is a direct contributory factor to the passage of cytokines through the BBB, and alcohol exerts pro-apoptotic effects on neuronal cells (Valles et al., 2004). Omega-3 fatty acids, N-acetyl cysteine, statins, coenzyme Q10, lipoic acid, curcuma, and TNF inhibitors are potential therapeutic neuroprotective factors acting on inflammatory, apoptotic, and oxidative pathways (Berk et al., 2011). Future treatments could include medications that act on neuroinflammatory pathways.
The roles of stress and trauma in immunity and autoimmunity offer new opportunities for research in patients with borderline personality disorder (BPD), post-traumatic stress disorder (PTSD), and traumatic brain injury. In BPD, the links with thyroid antibodies (Geracioti et al., 2003), increased pro-inflammatory cytokines (Kahl et al., 2006) and high rates (30–84%) of diffuse slow-wave and spike-wave EEG abnormalities (Boutros et al., 2003) raise the question of whether BPD is a form of chronic subacute encephalopathy that is related to autoimmunity. In PTSD, cortisol treatment has been proposed to ameliorate the intensity of intrusive memories and might also be useful in preventing low-grade systemic inflammation, which in turn, is discussed as a pathophysiological mechanism underlying somatic symptoms in PTSD through the cytokine pathway (Aerni et al., 2004; Rohleder et al., 2009). Traumatic brain and spinal injuries are associated with antibody production, which is responsible for a range of somatic symptoms that cannot be explained purely by neurological injury (Ankeny and Popovich, 2010).
Conclusions
The link between immune dysregulation, autoimmunity, and psychiatric illness may be closer than previously thought. The identification of aetiological markers to guide treatment has largely eluded psychiatrists. With the advent of sophisticated neuroimaging techniques and immunological markers, psychiatrists will be able to identify subsets of disorders that have an immunological basis. This review adds to the increasing body of evidence suggesting that immune dysregulation, neuroinflammation, and autoimmunity may play an important role in psychiatric disorders. NPSLE, anti-NMDA encephalitis, and EAAT are important differentials for the psychiatrist to consider when suspecting autoimmune encephalopathy. Even if the autoimmune disease is not directly aetiologically related to the psychiatric presentation, its detection is important due to the high morbidity and mortality associated with autoimmune diseases. Early intervention could result in a significant decrease in morbidity for patients and in cost savings for services by preventing multisystem involvement in the future.
In the end, it is clear that if the understanding of a disease is to be furthered in the 21st century, the current Cartesian mind–body dichotomy is an untenable paradigm, just as Plato discovered over 2000 years ago.
Footnotes
Acknowledgements
We would like to thank Professor David Castle and Professor Philip Boyce for their criticism and comments in the preparation of this manuscript.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Declaration of interest
SR is a founding trainer at Psych Scene, a company providing educational courses for psychiatry exams. SJH reports no conflict of interest. The authors alone are responsible for the content and writing of the paper.