
Abstract
Objective:
Bipolar disorder is a chronic, severe and disabling disease; however, its pathophysiology remains poorly understood. Recent evidence has suggested that inflammation and immune dysregulation play a significant role in the pathophysiology of bipolar disorder. This review is aimed to highlight the importance of systemic inflammation in modulating the inflammatory response of microglia and hence its potential involvement with bipolar disorder. We also discuss novel therapeutic strategies that emerge from this new research.
Method:
This article presents a theoretical synthesis of the effects of systemic inflammation on the immune response of the central nervous system in bipolar disorder. The complex relationship between stress, pro-inflammatory cytokines and microglial dysfunction is summarized, emphasizing the role of the kynurenine pathway in this process and, consequently, their effects on neuronal plasticity.
Results:
Bipolar patients demonstrate increased serum levels of pro-inflammatory cytokines (interleukin-1β, interleukin-6 and tumor necrosis factor-α) and lower hypothalamic–pituitary–adrenal axis sensitivity. This imbalance in the immune system promotes a change in blood–brain barrier permeability, leading to an inflammatory signal spread in the central nervous system from the periphery, through macrophages activation (M1 polarization). Chronic microglial activation can result in neuronal apoptosis, neurogenesis inhibition, hippocampal volume reduction, lower neurotransmitters synthesis and cytotoxicity, by increasing glutamate production and kynurenine metabolism.
Conclusions:
This review provides an overview of the mechanisms involved in the immune system imbalance and its potential involvement in the pathophysiology of bipolar disorder. Consequently, new strategies that normalize the immune-inflammatory pathways may provide a valuable therapeutic target for the treatment of these disorders.
Introduction
Bipolar disorder (BD) is a chronic, recurrent illness that represents a major public health concern and often shows incomplete recovery and increased mortality (Vieta et al., 2011). By 2020, BD is estimated to become the sixth leading cause of time lost due to disability or death among those aged 15–55 years (Gore et al., 2011). Most importantly, the disability related to BD is not only restricted to the symptomatic phases but also occurs during periods of remission (Rosa et al., 2014). Furthermore, poor functioning has been strongly associated with cognitive impairment in bipolar patients (Bonnín Cdel et al., 2014).
The natural history of BD progression involves relapses, persistent symptoms, comorbidities, cognitive impairment and neurobiological changes (Berk et al., 2011; Kapczinski et al., 2008). Post et al. (1992) suggested that multiple episodes lead to permanent alterations in neuronal activity, which may be transduced at the level of greater liability to relapse and poorer treatment response (Post et al., 1992). Therefore, episode frequency and severity, together with an augmented sensitivity to stress factors, may increase with the passing of time or with each new recurrence (Kapczinski et al., 2008; Post et al., 1992). However, little is known about the pathophysiological mechanisms involved in the neuroprogression of BD. Undoubtedly, a better understanding of these mechanisms might not only help to predict treatment response but also improve outcome measures, such as cognitive and psychosocial functioning.
Emerging evidence had shown that BD is accompanied by the activation of immune-inflammatory pathways as indicated by the increased levels of pro-inflammatory cytokines, positive acute-phase proteins, complement factors and increased levels of T-cell-related activation markers (Dargél et al., 2015; Rege and Hodgkinson, 2013). Immune disturbances have been associated with progression of the disorder evidenced by multiple episodes, longer duration of illness and medical comorbidities (Stertz et al., 2015). Nonetheless, systemic inflammation is directly related to macrophage activation and increased production of pro-inflammatory cytokines in the central nervous system (CNS). Th1 cytokines activate microglial enzyme tryptophan 2,3-dioxygenase (TDO) and indoleamine 2,3-dioxygenase (IDO), shifting microglial kynurenine (KYN) catabolism toward quinolinic acid (QUIN). This imbalance between QUIN and kynurenic acid (KYNA) synthesis promotes microglial susceptibility to stress which may be related to recurrence of new mood episodes and poor treatment response (Dantzer et al., 2008). In addition, BD is associated with disturbances in circadian rhythm and melatonin synthesis, facts that are related to the strength of CNS inflammatory signal (Etain et al., 2012).
This review summarizes the importance of systemic inflammation in modulating the inflammatory response of microglia and hence its potential involvement in the pathophysiology of BD. The authors also discuss novel therapeutic strategies that emerge from this new research.
Functioning of the hypothalamic–pituitary–adrenal (HPA) axis
Emerging evidence has shown the presence of an inflammatory profile in bipolar patients across all phases of the illness (Tsai et al., 2014). Compared to control subjects, patients with BD show an increase in the plasma concentration of interleukins (ILs) such as IL-1 receptor antagonist, soluble IL-2 receptor, soluble tumor necrosis factor (TNF) receptor 1 and C-reactive protein (CRP) (Tsai et al., 2014). Furthermore, an increase in IL-2 production and secretion is linked to activation of T-lymphocytes, more specifically, activation of Th1 lymphocytes, that exhibit inflammatory activity. The Th1 lymphocytes may trigger activation of the microglia, to activate the central inflammatory response and perform phagocytosis (Cherry et al., 2014). Recently, one study investigated inflammatory profile and functioning of HPA axis in patients with mood disorders. Their results showed that depressed men with lifetime hypomanic episodes had higher levels of CRP and cortisol than those without (hypo)mania (Becking et al., 2015). Thus, it has been proposed that cytokines secreted in chronic inflammation may cross the blood–brain barrier (BBB) and reach the cerebrospinal fluid (CSF), spreading the inflammatory signal to different regions of the CNS, including the HPA axis (Miller et al., 2013).
Increased levels of inflammatory cytokines can act by stimulating the HPA axis, as part of the physiological response to stress. The activation of the HPA axis is critical in situations of acute inflammation, with the purpose to fight infection through increased production of cortisol in humans, and corticosterone in rodents (Wright et al., 2005). However, in cases of chronic inflammation, the continued production of cytokines causes a reduction in expression and sensitivity of glucocorticoid receptors (GRs) in the hypothalamus and pituitary. The GRs are responsible for maintaining physiological negative feedback; therefore, in conditions of chronic stress, a reduction in this regulation is noted. Systemically, the reduction in GR sensitivity also inhibits anti-inflammatory response triggered by cortisol. Interestingly, the insensitivity of GR has been independently associated with BD (Fries et al., 2014). In addition, the use of a GR antagonist (mifepristone RU-486) has been shown to be effective in improving cognitive function in bipolar patients (Young et al., 2004). Taken together, HPA axis dysfunctions and elevated levels of pro-inflammatory cytokines may affect neuroplasticity with a negative impact on mood symptoms and cognition. Indeed, the lower sensitivity of the HPA axis as well as disturbances in the production of the corticotrophin-releasing hormone and adrenocorticotropic hormone have been related to a cortico-limbic dysfunction, with increased amygdala activity and less regulatory activity of hippocampus (HIP) in BD (Drevets et al., 2008). To understand how the HIP responds to glucocorticoids and how those changes are related to volume measures, Tata et al. (2006) carried out a study where male Sprague-Dawley rats were injected with corticosterone or vehicle for 60 days. They identified altered dendritic and glial processes as well as altered numbers and sizes of synapses on hippocampal CA3 (Tata et al., 2006). Other studies in rodents have shown that corticosterone administration decreases cell proliferation and survival in the dentate gyrus of HIP (Brummelte and Galea, 2010). Furthermore, it has been suggested that the inflammatory signal in the CNS, macrophages polarization and high-mobility group box 1 (HMGB1) secretion may be influenced by the concentration of cortisol and increased expression of GR (Frank et al., 2015; Weber et al., 2015). Finally, cortisol and chronic inflammation may affect the function of the CNS, increasing the enzyme TDO and IDO activity and reducing serotonin (5-hydroxytryptamine, 5-HT) synthesis (Maes et al., 2011).
Structure and functions of the BBB
The concept of BBB began at the end of the 19th century with Paul Ehrlich reporting that various dyes injected into the circulatory system were capable of staining all organs, but failed to stain the brain and spinal cord, leading to a hypothesis of the existence of two separated compartments (Patel and Frey, 2015). The BBB is composed of a monolayer of brain endothelial cells that form the walls of the capillaries, a basal membrane, pericytes, astrocyte end-feet and perivascular macrophages. Endothelial junctions like tight junctions and adherens junctions control cell permeability, resulting in a sealed structure (Abbott et al., 2010). The BBB regulates the exchanges between the CNS and the periphery, protecting the brain from toxic substances in the blood, supplying brain tissues with nutrients and filtering harmful compounds from the brain back to the bloodstream (Patel and Frey, 2015). Therefore, an intact and functional BBB is crucial to the maintenance of CNS homeostasis.
Besides the BBB, the CSF and meninges act as an anatomical protection to the CNS. This complex structure of layers surrounds the lymphatic system of CNS and avoids the entry of most stressors and pathogens into the brain. The meningeal lymphatic system located in the dura mater is responsible for draining the interstitial fluid, macromolecules and immune cells from the CNS to the lymph nodes (Louveau et al., 2015b). Interestingly, adaptive and innate immune cells have access and occupy meningeal spaces contributing to neuroimmune reactions (Louveau et al., 2015b). If a pathogen reaches the brain, a slow cascade of events occurs in order to remove the damaging antigen. First, the pathogenic antigen diffuses into the CSF and interstitial fluid via glymphatic system, through the vein walls and astrocytes end-feet (Louveau et al., 2015b). Consequently, those molecules are drained by the meningeal lymphatic vessels or by the lymphatic system of the nasal mucosa together with the immune cells and reach the cervical lymph nodes (Louveau et al., 2015a). At this point, this process should result in the onset of inflammatory response through the activation of adaptive immune cells, such as T-lymphocytes (Louveau et al., 2015a). This mechanism of clearance accounts to the immune privilege of the brain and is also important for the maintenance of homeostasis. However, if the peripheral inflammatory system is first activated, the cleaning process occurs much more quickly once the pathogen reaches the brain (Louveau et al., 2015a).
During chronic inflammation, toxins like lipopolysaccharide (LPS) and other pathogens enhance the release of pro-inflammatory cytokines, HMGB1 and other components that may increase microvascular permeability and lead migration of the leukocytes to the brain parenchyma (Frank et al., 2015; Weber et al., 2015). This infiltration triggers cytokines release and matrix metalloproteinases (MMPs) activation intensifying BBB disruption and inflammation in the CNS, through glia activation (Sumi et al., 2010). In fact, microglia activation and subsequent release of inflammatory mediators modulate the expression of adhesion molecules on endothelial cells that stimulates migration and recruitment of myeloid-derived blood cells to the brain. Therefore, reactive oxygen species (ROS) and cytokines (e.g. TNF-α and IL-1β) produced by activated microglia may impair BBB by altering the expression of molecules associated with endothelial cell junctions, such as claudin-5 and occludin, that are essential for its integrity (Da Fonseca et al., 2014).
Considering that inflammation plays a role in the neuroprogression of BD, as well as oxidative stress and microglial activation, Patel and Frey (2015) proposed a model of BBB dysfunction in BD. In this model, a persistent or transient loss of BBB integrity is associated with decreased CNS protection and increased permeability of pro-inflammatory mediators from peripheral blood into the brain, triggering microglial activation and promoting damage (Patel and Frey, 2015). In this regard, Zetterberg et al. (2014) assessed blood–CSF barrier function in 134 patients with BD and compared it with 86 healthy controls showing a significant increase in CSF/serum albumin ratio in BD patients (Zetterberg et al., 2014). Furthermore, it has been reported that mood stabilizers used for the treatment of BD, such as lithium and valproate, can inhibit MMPs function and attenuate BBB dysfunction (Yu et al., 2013).
Furthermore, it has been suggested that BBB may be vulnerable to changes in the microbiota and dysfunctions on intestinal barrier may contribute to the pathophysiology of psychiatric disorders. The intestinal barrier has an important role in the immune system modulation as it regulates the flow of external molecules or pathogens, but the primary function of this barrier remains in the absorption of nutrients, electrolytes and water from the diet (Kelly et al., 2015). The arrangement of the mucus layer, the microvilli epithelial cells layer and tight junctions contributes to intestinal functions and creates a barrier responsible for its protection (Kelly et al., 2015). However, the intestinal permeability can be easily altered by factors such as the diet, stressful events, alcohol ingestion and microbiota alteration (Berk et al., 2013). When the intestinal barrier is disrupted, bacteria from the gut can translocate to the lymph nodes and system circulation (Kelly et al., 2015). The LPS of the enterobacteria promotes the innate immune response, mainly through toll-like receptor-4 (TLR-4) activation, which triggers the release of pro-inflammatory cytokines and production oxidative stress compounds (Berk et al., 2013). Under stress conditions, this pro-inflammatory state could contribute to CNS immune activation, represented by TLR4 activation in the microglia cells and further increase the gut permeability (Berk et al., 2013; Kelly et al., 2015).
The link between gut microbiome and CNS seems to have implications in the pathophysiology of stress-related psychiatric disorders. Maes et al. (2013) found higher levels of immunoglobulin IgA and IgM and oxidative and nitrosative stress, against LPS, in chronically depressed patients compared to healthy individuals. These data suggest that an increased bacterial translocation may be a consequence of secondary systemic inflammation and also could act intensifying the primary inflammatory response in those individuals (Maes et al., 2013). Finally, a dysfunctional intestinal barrier could permit a microbiota-driven pro-inflammatory state with implications for the brain development, function and behavior and thus may contribute to the mood dysregulation.
Macrophage/microglia polarization
Macrophages are responsible for innate immune response activation in the periphery while microglia is the tissue-resident macrophages of the CNS, responsible for homeostasis and synaptic modulation (Nakagawa and Chiba, 2015). Microglia can be activated by damage-associated molecular patterns (DAMPs) as well as pathogen-associated molecular patterns (PAMPs) molecules through its receptors, such as TLRs family (Stertz et al., 2015). This activation triggers the expression of nuclear factor kappa B (NFkB), mitogen-activated protein kinase (MAPK) and interferon regulatory factor (IRF) signaling pathways that are accompanied by increased expression of pro-inflammatory genes. When acutely activated, microglia is considered an innate and adaptive mechanism that diminishes tissue damage and restores homeostasis (Nakagawa and Chiba, 2015). However, if the activation process extends for a longer period, the microglia produces higher amounts of TNF-α, IL-1β and HMGB1, leading to tissue inflammation, apoptosis and damaged neurotransmitter synthesis (Dantzer et al., 2008). It should be noted that HMGB1 can be released by other cell types (neurons and astrocytes) in stress situations, acting on TLRs and thereby mediating the inflammatory response and release of IL-1β by microglia (Frank et al., 2015). Furthermore, the release of pro-inflammatory cytokines promotes the BBB disruption—previously described—which could be an alternative for activated monocytes from peripheral blood to access the brain and thus exacerbating the neuroinflammation (Da Fonseca et al., 2014).
Since microglia exhibits significant plasticity, it can polarize into different phenotypes, depending on the subsets of transcription factors and secreted proteins that will define effector functions, such as killing or repair. Under normal conditions, microglia is responsible for CNS surveillance and also for the release of important neurotrophic factors (Franco and Fernández-Suárez, 2015). However, in the presence of an inflammatory stimulus, such as LPS, interferon-γ (IFN-γ) and TNF-α (Franco and Fernández-Suárez, 2015), a proinflammatory profile with killing activity, named M1, is induced. As a consequence of M1 polarization, several reactive oxygen and nitrogen species, newly synthesized pro-inflammatory cytokines (IL-1β and IL-6), chemokines (monocyte chemoattractant protein 1, MCP-1) and co-stimulatory molecules and receptors (MHC-II, CD32, CD80, CD86 and CD68) are expressed or up-regulated (Cherry et al., 2014; Franco and Fernández-Suárez, 2015). For example, certain IRFs such as IRF5 and IFR7, expressed under M1 polarization, activate the transcription of pro-inflammatory genes and suppress the expression of anti-inflammatory cytokines, such as IL-10 (Ferrante and Leibovich, 2012). After TLRs activation, those factors bind to an adaptor protein, the MyD88, leading the signaling cascade to the activation of NFkB and activator protein-1 (AP-1) and, consequently, the production of pro-inflammatory cytokines (Negishi et al., 2005).
At physiological conditions, a harmful event can be avoided by an M2 antagonist polarization, which is considered a resolving or repair anti-inflammatory phenotype (Nakagawa and Chiba, 2015). The M2 polarization is divided into three groups: M2a, M2b and M2c. When M2 is activated by IL-4 and IL-13, the phenotype is defined as M2a and has potent anti-inflammatory properties. Particularly, this phenotype exhibits up-regulation of arginase, inhibition of NFκB isoforms and enhanced expression of CD163, CD206 and phagocytic receptors (Duluc et al., 2007). Although the consequences of M2b polarization are still unknown, this phenotype is activated under TLRs stimulation by LPS or immune complexes (Nakagawa and Chiba, 2015). M2c is activated by IL-10 and transforming growth factor-β (TGF-β) and its function is related to tissue remodeling and extracellular matrix deposition after inflammatory process attenuation (Cherry et al., 2014). Anti-inflammatory ILs, such as IL-10, released by M2 phenotype, can also down-regulate M1 functions (Nakagawa and Chiba, 2015). IRFs like IRF4 may exert a negative control in the TLRs activation, by competing with IRF5 for MyD88 interaction, inducing the expression of M2 specific genes, such as arginase 1, and inhibiting the expression of pro-inflammatory genes dependent on IRF5 (Ferrante and Leibovich, 2012; Negishi et al., 2005).
In spite of this, it is important to consider that the current lack of knowledge about specific markers that distinguish resident microglia from circulating blood-derived macrophages in the human brain represents a limitation for cell-specific targeting of microglia in psychiatry. In this regard, Durafourt et al. (2012) compared polarization properties of human adult microglia and blood-derived macrophages in vitro showing a greater phagocytic activity in microglia than in macrophages. The expression of cell surface markers—such as CD23, CD163 and CD206—was only observed in M2 peripheral macrophages. Regarding M1 phenotype, microglia released higher levels of IL-10 than macrophages, while under M2 polarization, both cells produced IL-10 equally (Durafourt et al., 2012). Other researchers have shown that M1 response in peripheral macrophages may be activated by Th1 cells, while the M1 response from microglia could be self-activated through autocrine and paracrine mechanisms (Cherry et al., 2014). Although there is no validated method or marker for microglia and peripheral macrophage differentiation in the brain parenchyma, they may coexist in the CNS under some pathological situations (Franco and Fernández-Suárez, 2015).
Microglial dysfunction in BD
More recently, microglial dysfunctions have been the subject of study in the different psychiatric diseases (Watkins et al., 2014). In a qualitative post-mortem research, Bayer et al. (1999) found increased microglial activation associated with HLA-DR expression—an MHC-II—in the prefrontal cortex (PFC) of affective disorder and schizophrenia patients at later stages, when compared to healthy individuals (Bayer et al., 1999). Interestingly, Steiner et al. (2008) found a marked microgliosis in the PFC and thalamus of patients who committed suicide, suggesting a strong association between microglial activation and suicide in psychiatric patients (Steiner et al., 2008). Rao et al. (2010) found a significant increase in c-Fos, inducible nitric oxide synthase messenger RNA (mRNA), protein and mRNA levels of IL-1β and IL-1R, and components of the NFκB, in the post-mortem PFC from BD patients (Rao et al., 2010).
Importantly, despite suffering the influence of the inflammatory component, few in vivo studies have evaluated the activation of microglia in individuals with BD. Haarman et al.’s (2014) was the first report to demonstrate the activation of microglia in the HIP of bipolar patients compared with healthy individuals, through an increase in the uptake of gamma rays with the microglia-specific [(11) C]-(R)-PK11195 probe (Haarman et al., 2014). In the CSF of euthymic patients, Jakobsson et al. (2015) showed higher levels of MCP-1 and chitinase-3-like protein 1 (YKL-40), a glial activation biomarker, compared to healthy subjects. Higher levels of soluble CD14 and YKL-40 were also observed in the serum of these patients (Jakobsson et al., 2015). Additionally, higher levels of YKL-40 and MCP-1 observed in the CSF of BD patients suggest an inflammatory state, which may explain, in part, poor executive function and outcome (Rolstad et al., 2015).
As in vivo experiments evaluating microglial dysfunctions in psychiatric disorders are scarce, preclinical studies are a useful strategy to investigate the relationship between immune system alterations and neurological diseases. Even though there is no ideal model of mood disorders, most of them present certain similarities regarding pathophysiology or etiology of the disorder and treatment response. For instance, systemic immune activation by LPS administration may promote neuroinflammation and depressive-like behavior in mice, and these effects seem to be attenuated by chronic antidepressant administration (O’Connor et al., 2009). Using this model, Biesmans et al. (2013) observed a sickness behavior and mild depressive-like behavior associated with an increase in the IL-1β, IL-6, TNF-α and IFN-γ levels in the serum and in the brain of treated animals. Also, in the LPS group, the authors observed increased glial activation expressed by glial fibrillary acidic protein and ionized calcium binding adaptor molecule-1 (Iba-1) immunoreactivity, when compared to vehicle-treated mice (Biesmans et al., 2013). Dong et al. (2014) showed that lithium supplementation inhibited microglial activation and pro-inflammatory cytokines production, after LPS stimulation, in primary culture of microglial cells. According to the authors, these effects of lithium may be mediated by the PI3K/Akt/FoxO1 pathway activation (Dong et al., 2014).
Chronic unpredictable mild stress (CUMS) is probably the most widely used animal model of depression. With this model, Pan et al. (2014) detected increased levels of IL-1β in the serum, CSF and PFC of stressed animals. As expected, the expression of glial markers investigated in this study—such as CD11b for activated glia and Iba-1 for activated microglia—was increased in the PFC of rats submitted to CUMS, and these effects were reversed by the administration of antidepressants (Pan et al., 2014). Furthermore, the model of mania induced by d-amphetamine is well established and, besides the characteristic hyperlocomotion, there are evidence that treated animals show increased IL-4, IL-6, IL-10, TNF-α and carbonyl levels in the PFC, striatum and serum (Valvassori et al., 2015). Additionally, in the Valvassori’s study, lithium administration reversed d-amphetamine hyperactivity and decreased pro-inflammatory cytokine levels.
N-acetylserotonin/melatonin in BD
Sleep and biological rhythms are involved in the neurobiology of BD. Indeed, changes in the sleep–wake cycle, such as decreased need for sleep or insomnia/hypersomnia, are part of the diagnostic criteria for BD. Sleep/wake disturbance has been observed during mood states and even in periods of remission (Pinho et al., 2016). Sleep patterns and circadian rhythms are influenced by melatonin, a neurohormone synthesized primarily in the pineal gland during the dark phase of the night. The synthesis of melatonin involves the conversion of 5-HT to N-acetylserotonin (NAS) by the arylalkylamine N-acetyltransferase (AANAT) followed by the conversion of NAS to melatonin by the acetylserotonin O-methyltransferase (ASMT) (Etain et al., 2012).
More recently, researchers have investigated the relationship between immune-pineal melatonin and the innate immune response. The pineal gland is a target for PAMPs (e.g. LPS) or pro-inflammatory cytokines (TNF), and these signals can suppress the nocturnal release of melatonin by the gland, promoting the migration of leukocytes to the site of the lesion. Moreover, mononuclear and polymorphonuclear leukocytes can also synthesize melatonin. According to Muxel et al. (2012), during inflammatory response, macrophages induce the synthesis of melatonin, and that macrophage-synthesized melatonin may modulate the function of these professional phagocytes in an autocrine manner. In addition, the transcription factor NFkB seems to mediate PAMPs and pro-inflammatory cytokines-induced AANAT (the key enzyme involved in the synthesis of melatonin) in macrophages. These results indicate that the communication between pineal and extra-pineal sources of melatonin is dependent on NFkB nuclear translocation, which plays a significant role in the innate immune response, and that melatonin is part of this complex response.
Interestingly, Etain et al. (2012) assessed genetic and functional abnormalities of the melatonin biosynthesis showing that one frequent polymorphism located in the ASMT promoter, rs4446909, was associated with BD, with a lower mRNA level of ASMT and a lower enzymatic activity. These findings suggest that deleterious variants of ASMT might be associated with low 5-HT and NAS concentrations as well as melatonin level. Abnormalities of melatonin secretion may explain, in part, sleep/wake disturbance, abnormal actimetric parameters, and circadian preference evening observed in bipolar patients. Finally, sleep disturbance can also lead to changes in daily activities such as work, social activities and diet.
Nevertheless, Jang et al. (2010) proposed that the administration of NAS exerts antidepressant effect in mice during forced-swim test. It has been demonstrated that chronic treatment with fluoxetine increased the content of AANAT mRNA in the rat HIP, which suggests that NAS could be a mediator of the antidepressant action of drugs. The NAS, like brain-derived neurotrophic factor (BDNF), exerts its neuroprotective effect by the activation of tropomyosin receptor kinase B (TrkB), and this action seems to be independent of a neurotrophin or MT3 NAS receptor. Therefore, the NAS but not other 5-HT metabolites showed to have effects on synaptic plasticity, neurogenesis and synaptogenesis.
Taken together, these data support the concept of immune-pineal axis might be relevant to a better understanding of disorders with melatonin rhythms dysfunctions (e.g. BD) and might open new horizons for the treatment of psychiatric patients.
Cytokines effects on neuronal plasticity
Neurogenesis is the process by which neurons are generated from neural stem cells (Kim et al., 2016). Substantial production of new neurons in the adult brain mainly occurs in two limited areas: the subgranular zone in the hippocampal dentate gyrus and in the subventricular zone in the lateral ventricle (Kim et al., 2016). Neurogenesis consists of multiple steps, including neuronal precursor cell (NPC) proliferation, differentiation, survival, migration and integration into pre-existing hippocampal networks (DeCarolis and Eisch, 2010). Several studies have shown that adding new neurons into the pre-existing neural circuitry is crucial to the maintenance of neuronal plasticity and cognition in patients with psychiatric diseases, in particular BD (DeCarolis and Eisch, 2010).
Neuroimaging studies of BD have demonstrated abnormalities in neural circuits supporting emotion processing, emotion regulation and reward processing (Phillips and Swartz, 2014). Those neuroanatomic abnormalities include ventricular enlargement, gray matter loss in the HIP and cerebellum, volume decreases in the PFC and variations in the size of the amygdala (Roda et al., 2015). Although these changes become more pronounced with repeated episodes and longer duration of disease (Kapczinski et al., 2009), they have been demonstrated in individuals at risk of developing the disease (Phillips and Swartz, 2014). Furthermore, the reduced volume of amygdala and HIP in adults with bipolar depression tends to be normalized with lithium therapy (Hallahan et al., 2011). The volume changes in brain regions involved in BD—mainly atrophy—are thought to be the result of a reduction in the number of neuronal and glial density as well as the size and shape of the cell body (Uranova et al., 2004).
Pro-inflammatory cytokines may exert harmful effects on adult neurogenesis in the HIP. These cytokines are produced by activated microglia when the homeostasis of the microenvironment is disturbed, resulting in neuroinflammation. IL-1β is one of the major pro-inflammatory cytokines inhibiting adult neurogenesis (Koo and Duman, 2008). Progenitor cells in the subgranular zone have IL-1β receptors, which decrease cell proliferation when stimulated (Koo and Duman, 2008). Impairment of IL-1β action prevents the attenuated rate of adult neurogenesis in response to stress (Koo and Duman, 2008). In a recent study, Zunszain et al. (2012) reported that IL-1β-induced impaired neurogenesis may be reversed by a kynurenine 3-monooxygenase (KMO) inhibitor, which suggests the involvement of the kynurenine pathway (KYP) in the IL-1β regulation of hippocampal neurogenesis (Zunszain et al., 2012). Indeed, euthymic patients with BD showed an increase in blood KYN concentrations and KYN/tryptophan ratio (Reininghaus et al., 2014). Additionally, in patients with BD, higher levels of IL-1β were associated with dysfunction and suicide risk (Monfrim et al., 2014). Furthermore, when activated, microglia may promote expression of IFN-γ, which increases the activity of IDO followed by a rise in QUIN (Watkins et al., 2014), which causes excitotoxicity mediated by the N-methyl-D-aspartate (NMDA) receptor.
IL-6 and TNF-α are also pro-inflammatory cytokines associated with inhibition of adult neurogenesis. Monje et al. (2003) demonstrated that TNF-α and IL-6 decreased hippocampal neurogenesis in rats injected with LPS; such effects were completely blocked by systemic treatment with the nonsteroidal anti-inflammatory drug indomethacin (Monje et al., 2003). Another study from the same group showed that IL-6 induced depressive-like behaviors and impaired proliferation of hippocampal cells that may be mediated by NFκB signaling pathway (Monje et al., 2011). In addition, adult IL-6 knockout mice showed enhanced proliferation and survival of new neurons in the dentate gyrus and subventricular zone (Bowen et al., 2011). In the same line, Keohane et al. (2010) demonstrated that exposure of hippocampal NPCs to TNF-α during differentiation has a detrimental effect, suggesting that TNF-α may also inhibit neurogenesis (Keohane et al., 2010).
5-HT and KYN metabolism
One of the main neurotransmitters affected by inflammation is the 5-HT. The decrease in bioavailability of tryptophan and increase in KYN formation lead to a reduction in the synthesis of 5-HT, which may be related to the classic depressive symptoms (Myint, 2012). It is important to remember the biochemical basis involved in the synthesis of these two compounds to understand the influence of systemic inflammation on 5-HT metabolism and KYN in the CNS.
The deviation of 5-HT to KYN synthesis, through the action of IDO and TDO in the liver, astrocytes and microglia, has become an important etiologic factor for mood disorders (Watkins et al., 2014). In physiological situations, 1–5% of tryptophan is metabolized to 5-HT. Around 20–30% of the normal production of 5-HT takes place in the CNS, after the passage of systemic tryptophan through the BBB (Myint, 2012). An increase in KYN metabolism directly decreases the synthesis of 5-HT and melatonin. Melatonin formation is dependent on the metabolism of 5-HT to NAS. Melatonin secretion is bypassed in situations where an increase in the activity of IDO is observed (Oxenkrug, 2013). Therefore, melatonin biosynthesis is essential for maintaining the circadian cycle in humans, and the reduction in melatonin concentration causes disturbances in sleep and circadian rhythms, which are common signs in patients with neuropsychiatric disorders.
It is known that under normal physiological conditions, tryptophan metabolism is primarily maintained by the liver function. However, in systemic or central inflammation, the activity of IDO is increased in extrahepatic tissues, including the CNS (Heyes et al., 1993). The activity of IDO is stimulated by IFN-γ and TNF-α and is inhibited by the anti-inflammatory cytokine IL-4 (Myint and Kim, 2003). Under these circumstances, the activity of the KMO is increased, leading to a greater formation of 3-hydroxykynurenine (3-HK) and QUIN compared to the KYNA formation (Munn et al., 1999). As previously mentioned, QUIN is an agonist of NMDA receptors while KYNA is an NMDA receptor antagonist and, therefore, is protective against excitotoxicity of QUIN. This fine regulation of the KYN metabolism directly affects the availability of 5-HT in the CNS, and it may be related to the clinical signs observed in mood disorders.
It is well established that patients with mood disorders experienced increased pro-inflammatory cytokines such as IL-6, IL-2, TNF-α and IFN-γ (Myint, 2012). The release of inflammatory markers correlates positively with the IDO activity and decreases the availability of 5-HT, as previously mentioned. In addition, an imbalance between QUIN/KYNA syntheses promotes microglial susceptibility to stress. The increased sensitivity of microglia appears to be related to the recurrence of mood episodes and treatment resistance (Myint and Kim, 2003). The relationship between KYN and tryptophan reinforces the relevance of this metabolic pathway in this pathological context. In fact, bipolar patients presented higher serum levels of KYN and KYN/tryptophan ratio compared to control subjects (Reininghaus et al., 2014). Furthermore, higher TDO activity and KYN concentrations were observed in the anterior cingulate cortex from patients with BD compared to controls (Miller et al., 2006). Another post-mortem study showed an increase in the gene expression of tryptophan hydroxylase, the enzyme responsible for the conversion of tryptophan to 5-hydroxytryptophan, in the dorsolateral PFC of patients with BD (De Luca et al., 2005).
As mentioned, microglia may promote the production of QUIN and 3-HK during inflammatory conditions; both mediators can induce neuronal death, especially in the HIP and cortex (Chiarugi et al., 2001). In a morphometric magnetic resonance imaging study, a positive correlation between KYNA/QUIN ratio and the volume of gray matter in the HIP and amygdala of unmedicated patients with major depression was observed (Savitz et al., 2015). In BD, a strong correlation between KYNA/3-HK and hippocampal volume has also been demonstrated, suggesting a relationship between the KYN pathway and neuroprogression of BD (Savitz et al., 2015).
Emerging therapeutic strategies
New concepts of pathogenesis have shown that BD is associated with activation of immune-inflammatory pathways, which may lead to macrophage activation and pro-inflammatory cytokines production. These inflammatory signals increase BBB permeability allowing leukocytes migration through brain parenchyma with subsequent glial activation and damage augmentation. The microglia is mainly activated to M1 pro-inflammatory phenotype, which produces ROS—related to neuronal apoptosis—and more pro-inflammatory cytokines—responsible for neurogenesis impairment. Furthermore, pro-inflammatory cytokines stimulate HPA axis, decreasing GR expression and sensitivity and enhancing cortisol release. Both M1 activation and cortisol activate IDO/TDO leading to QUIN formation and reducing 5-HT synthesis. As an NMDA agonist, the QUIN stimulates glutamate release contributing to excitotoxicity, ROS production and, consequently, neuronal death (see Figure 1). Therefore, the search for new agents that can modulate the complex mechanisms involved in the pathophysiology of BD could represent a therapeutic opportunity for the treatment of these disorders.
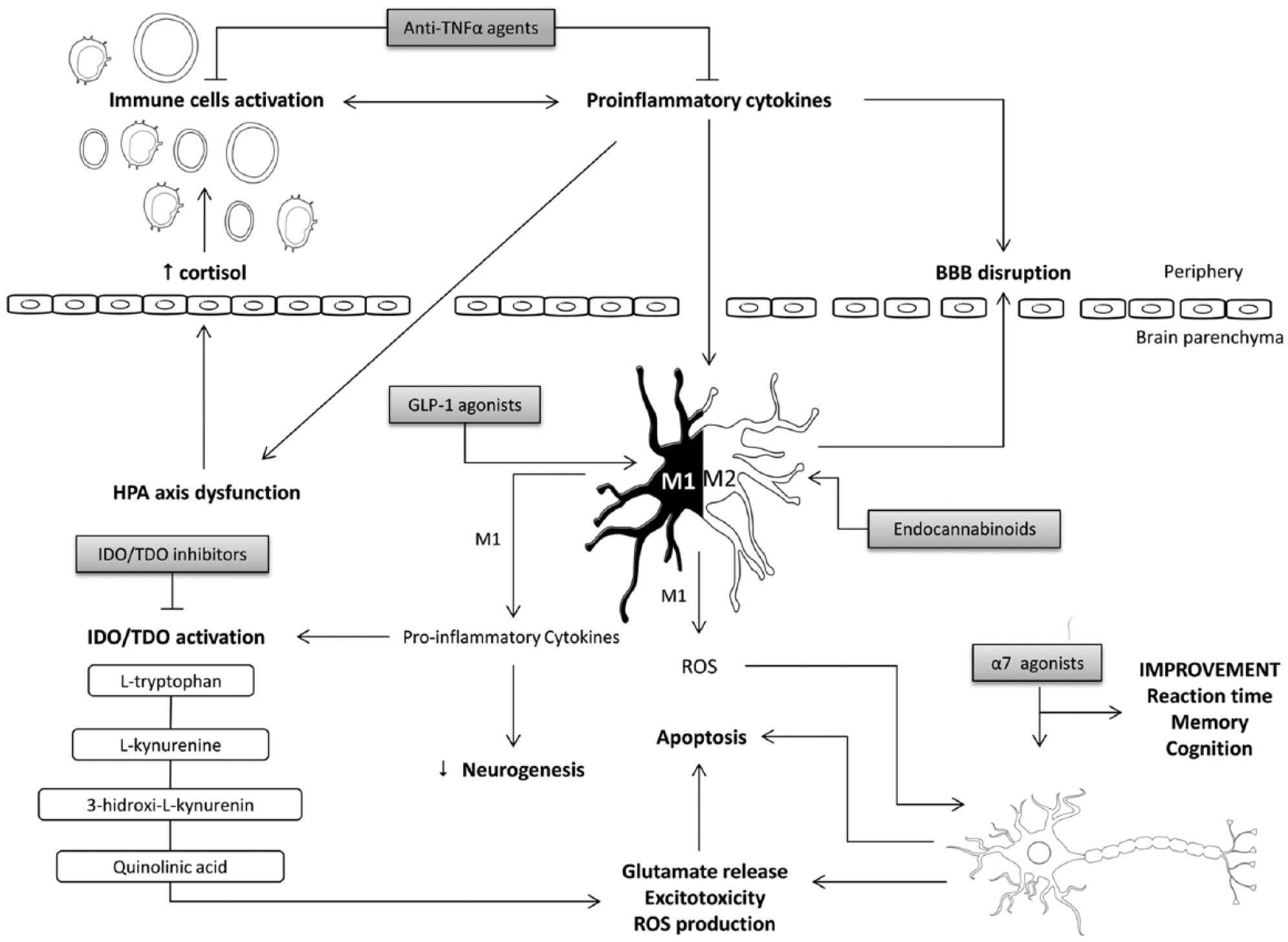
Activation of immune-inflammatory pathways in BD. Evidence suggests that BD is associated with inflammatory activation leading to macrophage activation and cytokines pro-inflammatory production. These inflammatory signals increase BBB permeability allowing leukocytes migration through brain parenchyma with subsequent glial activation and damage augmentation. The microglia is mainly activated to M1 pro-inflammatory phenotype which produces ROS—related to neuronal apoptosis—and more pro-inflammatory cytokines—responsible for neurogenesis impairment. Also, pro-inflammatory cytokines stimulate HPA axis, decreasing GR expression and sensitivity and enhancing cortisol release. Either M1 activation or cortisol activate IDO/TDO leading to QUIN formation and reducing 5-HT synthesis. As an NMDA agonist, the QUIN stimulates glutamate release contributing to excitotoxicity, ROS production and, therefore, neuronal death. Also, along the inflammatory pathway, the emerging therapeutic strategies are indicated accordingly to their site of action (gray boxes).
IDO inhibitors—1-methyl-D,L-tryptophan
Emerging evidence suggests that pro-inflammatory cytokines may influence 5-HT signaling in the brain and periphery through the KYP. Cytokines, such as IL-2 and IL-6, released in the periphery from macrophages and IFN-γ from activated microglia may promote overstimulation of the KYP that effectively decreases overall 5-HT availability in the brain as well as alters neurotrophic factors and TNF-α. IDO activity is enhanced by IFN-γ, consequently decreasing 5-HT CNS levels and increasing neurotoxic metabolite QUIN. Overproduction of QUIN may decrease BDNF, activate pro-apoptotic cascades by TNF-α and increase glutamate and NMDA receptor-activated excitotoxicity (Watkins et al., 2014). Changes in synaptic resilience, connectivity of serotonergic neurons and programmed cell death may occur as a consequence of the activation of this mechanism.
The inhibition of IDO activity may protect against neurotoxicity mediated by NMDA receptors. This hypothesis was tested in a model of Huntington disease where a chronic increase in expression and activity of IDO was followed by the production of neurotoxic metabolite 3-HK. In this model, IDO knockout mice exhibited lower sensitivity to QUIN-induced neurotoxicity in the striatum, when compared to control group. It suggests that IDO inhibitors may promote the best balance between QUIN and KYNA, protecting against excitotoxicity mediated by NMDA receptors (Mazarei et al., 2013).
In an LPS-induced-depressive-like behavior animal model, 1MT, an inhibitor of IDO, was shown to reduce the immobility time in the forced-swim test and tail suspension test. This behavioral improvement was correlated with a reduction in KYN/tryptophan ratio in the brain of mice (O’Connor et al., 2009). It is important to note that there are no clinical studies using 1MT in the psychiatric disorders.
In a chronic stress model, the co-treatment with allopurinol, an inhibitor of TDO activity, also attenuated the immobility time in the forced-swim test; this effect was correlated with reduced circulating KYN concentrations (Gibney et al., 2014). Furthermore, an increase in TDO activity was observed in post-mortem brain of patients with schizophrenia and BD and seems to be related to the increase in CNS metabolites of KYN (Miller et al., 2008). Recently, the benefits of allopurinol were also demonstrated in patients with BD (Jahangard et al., 2014).
Taken together, these evidences show that the M1 phenotype and inflammatory cytokines produced by these cells act on KYN metabolism, leading to an increase in toxic metabolites (QUIN) and, consequently, neurotoxicity. Although IDO inhibitors do not directly modify macrophage/microglia polarization toward M1 or M2 phenotype, these agents could modulate the KYP decreasing the production of toxic metabolites and promoting 5-HT secretion. Probably, this mechanism may contribute to the improvement of mood symptoms demonstrated in some preclinical studies.
Anti-TNF-α agents
TNF-α is an 185-aminoacid glycoprotein that was initially described for its ability to induce necrosis in certain tumors. TNF-α is mainly produced by monocytes and macrophages or microglia in the CNS. It is a potent inducer of the inflammatory response, a critical regulator of innate immunity and plays an important role in the regulation of Th1 immune responses against pathogens (Karson et al., 2013; Miller et al., 2013). Given that BD is associated with a low degree of inflammatory state, it is plausible to speculate that anti-TNF-α agents could modulate this process and be a useful therapeutic strategy for this illness.
Şahin et al. (2015) showed that animals that were treated with the commercially available chimeric monoclonal antibody against TNF-α, infliximab, experienced lower cognitive impairment and higher BDNF levels compared to controls in a model of chronic stress (Şahin et al., 2015). Likewise, chronic treatment with anti-TNF-α decreased depressive-like behavior and anxiety symptoms in the same experimental model (Karson et al., 2013). Clinically, Raison et al. (2013) showed that depressed patients treated with infliximab plus antidepressants presented higher improvement in depressive symptoms than controls. This effect was particularly relevant for patients who had serum CRP levels higher than 5 mg/L than others (Raison et al., 2013). This study was a first step toward individualizing treatment for patients with mood disorders by showing that individuals with a pro-inflammatory profile could benefit from therapy with biological agents.
Furthermore, Kroner et al. (2014) assessed the effects of TNF-α on macrophage polarization in a model of spinal cord injury. They showed that wild-type mice experienced greater M2/M1 ratio when compared to TNF knockout mice, as evidenced by a two-fold increase in the M2 markers arginase-1 and CD206. It suggests that the expression of TNF may contribute to the predominantly M1 cytotoxic macrophages (Kroner et al., 2014).
To sum up, studies have demonstrated the influence of TNF-α on Th1 lymphocytes in promoting a release of pro-inflammatory cytokines or inducing the M1 polarization highlighting the potential use of anti-TNF-α as a modulator of peripheral or central immune system dysfunctions.
Glucagon-like peptide-1 receptor agonists
Glucagon-like peptide-1 (GLP-1) is a hormone released after food ingestion that facilitates insulin release from pancreatic cells (Hölscher, 2014). As GLP-1 has a short half-life, some commercially available analogs such as liraglutide and exenatide that are resistant to degradation by the dipeptidyl peptidase 4 were developed and had been used for the treatment of type 2 diabetes (Hölscher, 2014).
However, glucagon-like peptide-1 receptors (GLP-1Rs) are not exclusive of the pancreas and have been observed in other tissues including the brain. GLP-1R agonists exert neuroprotective effects related to cell growth and repair, inhibition of apoptosis and reduction of inflammatory responses (Isacson et al., 2011; Kim et al., 2009). For instance, astrocytes and microglia induce the GLP-1R expression, and the treatment with GLP-1 agonists prevents the expression of inflammatory markers (NFκB and IL-1β) in vitro and in vivo (Hölscher, 2014).
The role of GLP-1 analogs has been suggested as a promissory therapeutic strategy for cognitive dysfunction and neurodegenerative disorders. For instance, Kim et al. (2009) found that exendin-4, an analog of GLP-1, prevented 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced microglial activation in the substantia nigra and striatum, in a rodent model of Parkinson’s disease. In this study, exendin-4 suppressed the MPTP-induced expression of TNF-α and IL-1β. Additionally, dopaminergic neuron death in the substantia nigra was significantly reduced 1 week after MPTP administration in exendin-4-treated animals, emphasizing the protective effects of exendin-4 in the CNS.
Although the evidence assessing GLP-1R agonists in psychiatry are scarce, Isacson et al. (2011) showed that rats and mice treated with exendin-4 (during 1–2 weeks) had an improvement in anxiety and depressive symptoms when compared to controls and imipramine-treated animals. Additionally, they observed an increase in the hippocampal neurogenesis in those animals treated with exendin-4 chronically, which was correlated with the improvement during the behavior tasks. Further understanding of the distinct pathways involved in the neurogenesis may help reveal how GLP-1 analogs exert mood regulation.
Agonists of the nicotinic alpha-7 receptor (α7 nAChRs)
Several lines of evidence have shown that cholinergic anti-inflammatory pathway may be directly involved in the pro-inflammatory cytokines release. The α7 nAChRs are expressed on the surface of macrophages as well as in neurons and microglial cells of the mammalian brain. In a mouse ischemic stroke and bone fracture model, Han et al. (2014) showed that the anti-inflammatory activity of α7 nAChRs agonists was mediated via normalization of imbalance microglia/macrophage polarization states. In particular, α7 nAChRs agonist treatment reduced microglia/macrophage activation, decreased M1 microglia/macrophages and increased M2 macrophages as well as increased anti-oxidant enzymes. Likewise, Li et al. (2011) found that LPS-induced TNF-α release was inhibited by A-833834, a high affinity and selective α7 nAChR agonist, in both mouse peritoneal macrophages and human whole blood in vitro; such effects were attenuated by α7 nAChR antagonist. In addition, intraperitoneal administration of A-585539, another α7 nAChR agonist with limited brain penetration, decreased LPS-induced TNF-α release in mouse serum.
The use of α7 nAChRs agonists in psychiatry has recently gained attention, and their use has shown positive effects on cognition in preclinical (McLean et al., 2012) and clinical studies (Olincy and Freedman, 2012). McLean et al. (2012) demonstrated that PNU-282987, a selective α7 nAChRs agonist, can reverse the cognitive deficit induced by phencyclidine in an animal model of schizophrenia. More recently, a double-blind, randomized, placebo-controlled study was conducted to test the efficacy of encenicline, a selective α7 nAChRs agonist, for the treatment of cognitive impairment in schizophrenia (Keefe et al., 2015). A decrease in mean Schizophrenia Cognition Rating Scale (SCoRS) total scores was observed over time, and there was a significant difference for encenicline 0.9 mg vs placebo (Keefe et al., 2015).
As far as we know, the role of α7 nAChRs agonists treatment was not investigated in BD. However, the fact that these agents showed to reduce inflammation and oxidative stress, which are often associated with the pathophysiology of illness progression, may suggest that activation of α7 nAChRs could represent a therapeutic opportunity for the treatment of psychiatric patients.
Endocannabinoids
This system is an interesting possible target for modulating neuroinflammation due to the fact that the expression of the cannabinoid receptor type 2 (CB2) is low in resting microglia but is considerably high in activated microglia (Franco and Fernández-Suárez, 2015). In a model of traumatic brain injury, Tchantchou et al. (2014) showed that an inhibitor of anandamide (arachidonoyl ethanolamide) hydrolysis, an endogenous ligand to endocannabinoid, was capable of modulating microglial phenotype toward the M2 type, as evidenced by an increase in the arginase-1 activity (Tchantchou et al., 2014). Consistent with these results, in a recent study, Mecha et al. (2015) demonstrated that M2 polarization occurs upon exposure to anandamide and 2-arachidonoyl glycerol in microglial cultures (Mecha et al., 2015). Moreover, this study showed that an antagonist of cannabinoid receptor blocked M2 polarization, and that CB2 knockout mice have suppressed M2 polarization (Mecha et al., 2015). Given that the endocannabinoid machinery has an important role in the regulation of microglial activation, further studies should investigate the benefits of this pathway in psychiatric disorders.
Conclusion
This review provides a synthesis of the consequences of systemic inflammation in the immune response of the CNS in BD and discusses possible mechanisms involved in this process. M1 and M2 polarization states of macrophages play a significant role in shifting the immune response toward a pro-inflammatory or anti-inflammatory response. The biased pro-inflammatory milieu observed in BD patients may promote an increased permeability of the BBB, leading to a massive recruitment and infiltration of inflammatory markers from the periphery into the brain, triggering microglial activation and proliferation. Like M1 peripheral macrophages, M1 polarized microglia promotes the secretion of inflammatory cytokines, amplifying the inflammatory response, resulting in neural network dysfunctions with consequent impact on mood symptoms, cognition and treatment response. Consequently, new strategies that normalize the immune-inflammatory pathways may provide beneficial therapeutic opportunities for the treatment of BD.
Footnotes
Acknowledgements
Luiza P Géa and Florência M Barbé-Tuana are recipients of scholarship from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq). Bruna M Ascoli is a scholarship recipient from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES). Bruna M Ascoli and Luiza P Géa contributed equally to this work.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Bruna M Ascoli, Luiza P Géa, Rafael Colombo, Florência M Barbé-Tuana and Adriane R Rosa declare no possible conflicts of interest, financial or otherwise, or grants or other forms of financial support. Flávio Kapczinski has received grant/research support from AstraZeneca, Eli Lilly, Janssen-Cilag, Servier, CNPq, Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), NARSAD and Stanley Medical Research Institute; has been a member of the board of speakers for AstraZeneca, Eli Lilly, Janssen and Servier; and has served as a consultant for Servier.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.