
Abstract
Introduction: FTLD as a selective neurodegeneration resulting in neural circuit dysfunction
Nature uses only the longest threads to weave her patterns, so that each small piece of her fabric reveals the organization of the entire tapestry.
The frontotemporal lobar degenerations (FTLD) have onset typically in the sixth and seventh decades, and as such are devastating for mature adults, leading to a rapid decline and resultant early death, often under a decade from diagnosis (Mocellin et al., 2008). Thus, early diagnosis and development of reliable biomarkers to assess the progress of the illness are crucial for patients and their families to both stage and plan for difficulties that will arise due to cognitive decline, and to develop innovative treatments.
Recent neuropathological and neuroimaging studies of FTLD have converged upon the investigation of neural circuits or networks (Mesulam, 2009; Seeley et al., 2008, 2009). The fronto-striato-pallido-thalamo-cortical re-entrant neural circuits (Cummings, 1993) (Figure 1) are subsystems of large-scale neural networks under investigation as structural–functional substrates for the clinical features of FTLD and in particular, the frontotemporal dementia (FTD) subtype. Neuroplastic structural change in the frontostriatal circuits may manifest as morphological change. Network disruption may arise from loss of function from any node within the circuit. Key nodes that may be disrupted by neuropathological change are the frontal lobes and the striatum, together with the interconnecting white matter. Similarly, thalamus and globus pallidus may be affected. Within frontostriatal circuits, the neostriatum, comprising the caudate nucleus and putamen, occupies a central strategic role as a neural relay centre, with specific linkages to frontal and parietal cortex (Draganski et al., 2008). While specific morphological and functional change of the cortical regions, and to a lesser extent white matter, have been extensively investigated in FTD, there has been less interest in the striatum. However, the specific interconnections of the striatum to frontal and related cortex may allow the striatum to serve as a miniature map of the corresponding morphological change in other nodes of the frontostriatal circuits. Accordingly, the specific morphology of the caudate nucleus and putamen may correspond to morphological, and hence functional change, in the neural circuits relayed. It is acknowledged that structural and functional changes in cortex and white matter may represent the primary changes in FTD; however, the measurement of such morphological change as a clinically useful biomarker is likely to remain elusive due to the inter-individual variation in frontal cortical morphology, and the relative complexity of white matter morphology in FTD. Disruption of any component of the frontostriatal circuits will potentially affect network functions, and such network disruption may have up- and downstream effects due to trans-synaptic neurodegeneration (Buren, 1963). Thus, it is possible that circuit-based dysfunction and morphological change may be reflected, at least in part, in all nodes of the circuits (Looi, 2011). Given that the striatum occupies a strategic nodal location, and the close correspondence of striatal structure to its interconnections, the striatum may be a key structure in which morphological change may be detectable (Looi, 2011).
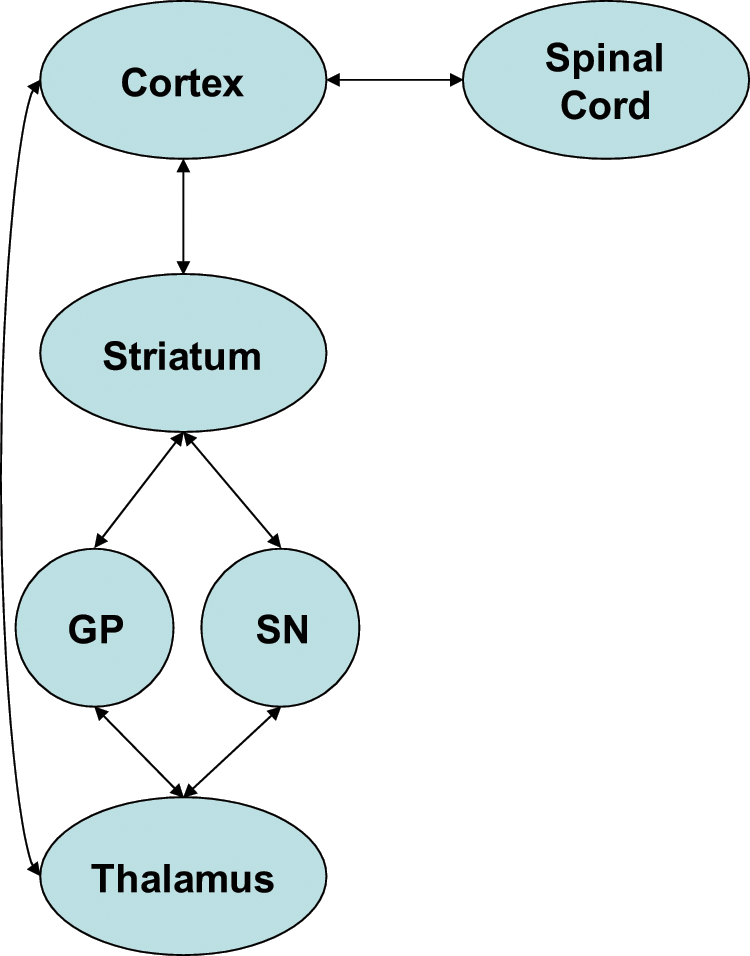
Frontostriatal circuits.
There is burgeoning neuropathological and neuroimaging evidence that the striatum is affected early and significantly in FTLD, with differentiation based upon subtype due to relative degrees of frontostriatal circuit involvement (Chow et al., 2008, Garibotto et al., 2011; Looi et al., 2008, 2009, 2010). Striatal atrophy may thus be an early biomarker of FTD, and a harbinger of frontostriatal circuit neurodegeneration (Looi et al., 2010, 2011a). The frontostriatal circuits comprise pathways involved with cognitive, emotional, behavioural and motor functions. Accordingly, dysfunction may be manifest in these domains, and can be observed clinically. Our morphological studies, together with similar studies by other groups, have demonstrated that caudate, and to a lesser extent, putaminal atrophy is characteristic of FTLD compared to Alzheimer’s disease and controls (Chow et al., 2008, Garibotto et al., 2011; Looi et al., 2008, 2009, 2010). In FTLD, we have identified general and specific patterns of morphological change in the neostriatum with functional implications for frontostriatal circuits (Looi et al., 2010, 2011a). The combination of observable clinical manifestations, along with the morphology of the striatum as a biomarker of frontostriatal circuit structural integrity, constitutes a basis for a potential endophenotype. Thus FTD may, in part, be reconceptualised as a frontostriatal disorder.
Current diagnostic criteria for FTLD: Limitations and challenges
The existing consensus criteria for FTLD focus upon clinical features of illness, and patterns of impairment (Neary et al., 1998), reflecting the concept that FTLD represents a super-ordinate clinical syndrome encompassing several different neurodegenerative diseases (Cairns et al., 2007), with characteristic clinical subtypes. There are three main clinical subtypes of FTLD: behavioural variant frontotemporal dementia (FTD), progressive-non-fluent aphasia (PNFA), and semantic dementia (SD). FTD is characterised by greater frontal-system dysfunction behavioural change manifest as early decline in interpersonal interaction/conduct, loss of insight and emotional blunting; SD and PNFA are believed to represent predominant temporal atrophy with specific impact upon language function (Neary et al., 1998). The relationship between clinical subtype and neuropathology remains problematic, due to the fact that the same clinical features may arise from different neuropathological processes. However, there may be commonalities in the pattern of neurodegeneration which bear a closer relationship to clinical features. Neural circuit dysfunction may form a useful framework to understand such consequences of neurodegeneration. Such dysfunction would potentially be associated with specific clinical features based upon the pathways involved, and the functions they subserve. Thus, study of the neurodegeneration of structural components of neural circuits may prove fruitful in FTLD. Magnetic resonance imaging (MRI) affords a means of studying brain structure in vivo. Measurement of structural change in the brain using MRI allows potential visualisation of neurodegenerative morphological change, or at least the component manifest as atrophy, and associated changes in vivo. However, we acknowledge neurodegenerative processes may precede any identifiable morphological change, and in other instances may not manifest as morphological change.
To date, research in FTLD has focused on characterisation of the clinical patterns of decline and upon cortical neuropathology. Indeed, the clinical consensus criteria for FTD describe essentially behavioural changes ascribed to atrophy of frontal and temporal cortical regions. This results in a certain degree of circularity. Because the clinical features are predicated on a cortical conceptualisation of FTD, there is a selection for clinical samples that fulfil such criteria. Similarly, the macroscopic pathology and neuroimaging of FTD has focused upon focal atrophy of frontal or temporal lobes (Schroeter et al., 2007), whilst microscopic pathology has focused upon the molecular pathology of neuronal loss and gliosis (Cairns et al., 2007) with some neuroimaging interest in white matter disease in FTD (Zhang et al., 2009), and there has been no specific investigation of subcortical structures.
Whilst these are necessary stages in the evolution of the understanding of FTD, it is also possible to attempt to understand FTD on the basis of distributed neural networks, in which dysfunction due to aggregation of misfolded proteins in vulnerable cell populations may result in neurodegenerative disease affecting such networks (Seeley et al., 2006, 2008, 2009). In their 2008 study, Seeley et al. used voxel-based morphometry of MRI to suggest that atrophy occurred in the frontal paralimbic network in early behavioural-variant FTD, comprising associated atrophy of dorsolateral and polar frontal cortex, dorsal insula, thalamus, hippocampus and the striatum (Seeley et al., 2008). Whilst such studies comprise investigation of large-scale distributed brain networks (Mesulam, 2009), there exists a smaller, more constrained ensemble of circuits: the frontostriatal circuits, which are of burgeoning interest in FTD as their dysfunction may also be linked to the pattern of deficits seen in this disease group.
Frontostriatal circuits and FTD: The central role of the neostriatum
The neostriatum is a critical relay in frontostriatal circuits, as it comprises specific re-entrant interconnections among frontal, striatal, pallidal and thalamic regions (Alexander et al., 1986, Draganski et al., 2008). The cognitive and behavioural abnormalities identified in FTD may be conceptualised in terms of dysfunction in frontostriatal circuits (Hodges and Patterson, 2007). Frontostriatal circuits or loops include a discrete prefrontal region which sends efferent pathways through the neostriatum (caudate nucleus, putamen) or nucleus accumbens, via the globus pallidus, onto the thalamus, and thence to the back to that specific prefrontal cortex (Alexander et al., 1986). These loops include motor loops originating in the frontal eye fields and supplementary motor cortex, as well as ‘cognitive loops’ arising from dorsolateral prefrontal cortex, anterior cingulate cortex and orbitofrontal cortex (Alexander et al., 1986). Circuit dysfunction may result in characteristic cognitive and behavioural syndromes (Cummings, 1993).
The dorsolateral prefrontal circuit (DLPFC), arising from Brodmann areas 9 and 10 (Figure 2), mediates problem solving, verbal/non-verbal fluency and retrieval from memory, and is linked to the limbic memory system. Clinical syndromes associated with DLPFC dysfunction are described as executive dysfunction and are characterised by deficits in cognition related to the above domains (Cummings, 1993; Koziol and Budding, 2009). The orbitofrontal circuit (OFC), arising from Brodmann areas 10 and 11 (Figure 3), mediates inhibition and impulse control. Clinical syndromes associated with OFC dysfunction are described as emotional and social dysfunction and are characterised by deficits in social judgement and impulse control (Cummings, 1993; Koziol and Budding, 2009). Recently, subdivisions of the OFC into lateral and medial portions have been described, with lateral Brodmann areas 10 and 11 connecting to ventromedial caudate, and medial Brodmann area 11 connecting to the nucleus accumbens (Tekin and Cummings, 2002). The anterior cingulate (or ventromedial prefrontal) circuit (ACC), arising from Brodmann area 24 (Figure 4), mediates motivation and initiation of behaviour. Clinical syndromes associated with ACC dysfunction are described as apathy and loss of motivation, including akinetic mutism, and are characterised by lack of motivation (Cummings, 1993; Koziol and Budding, 2009). Therefore, syndromes associated with frontostriatal circuit dysfunctions are consistent with many of the clinical manifestations of FTD.
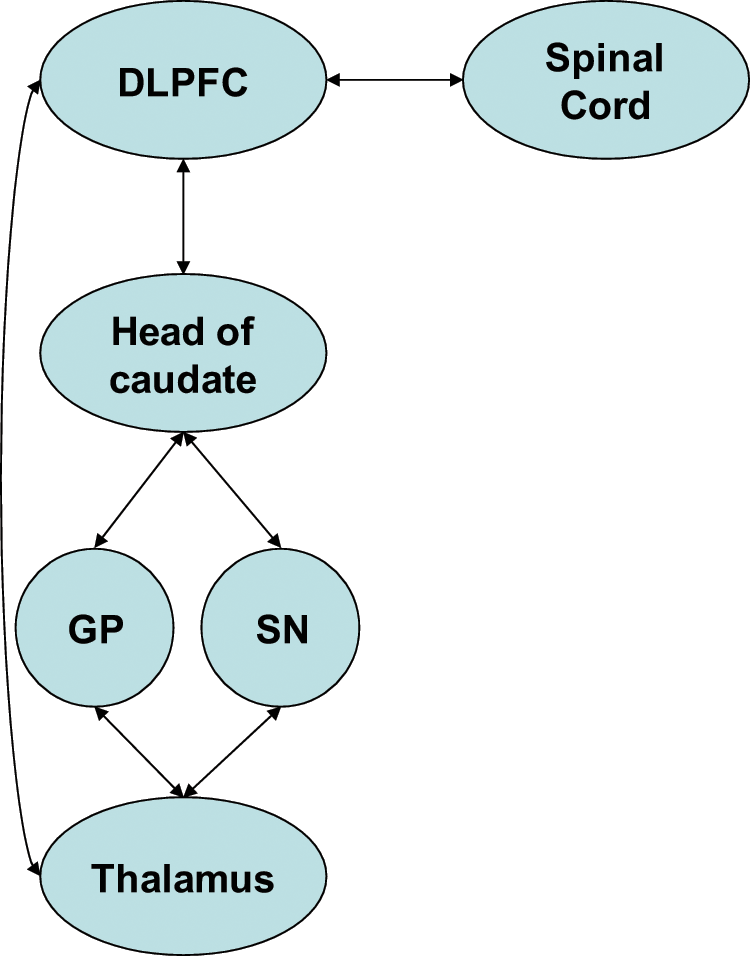
Dorsolateral prefrontal cortex circuit.
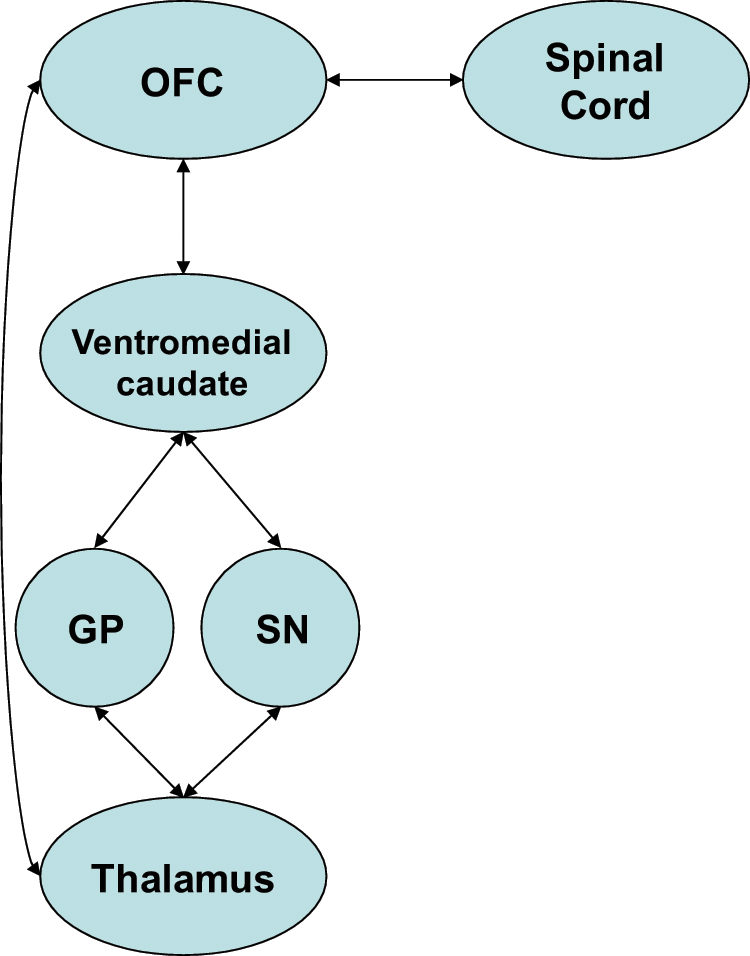
Orbitofrontal cortex circuit.
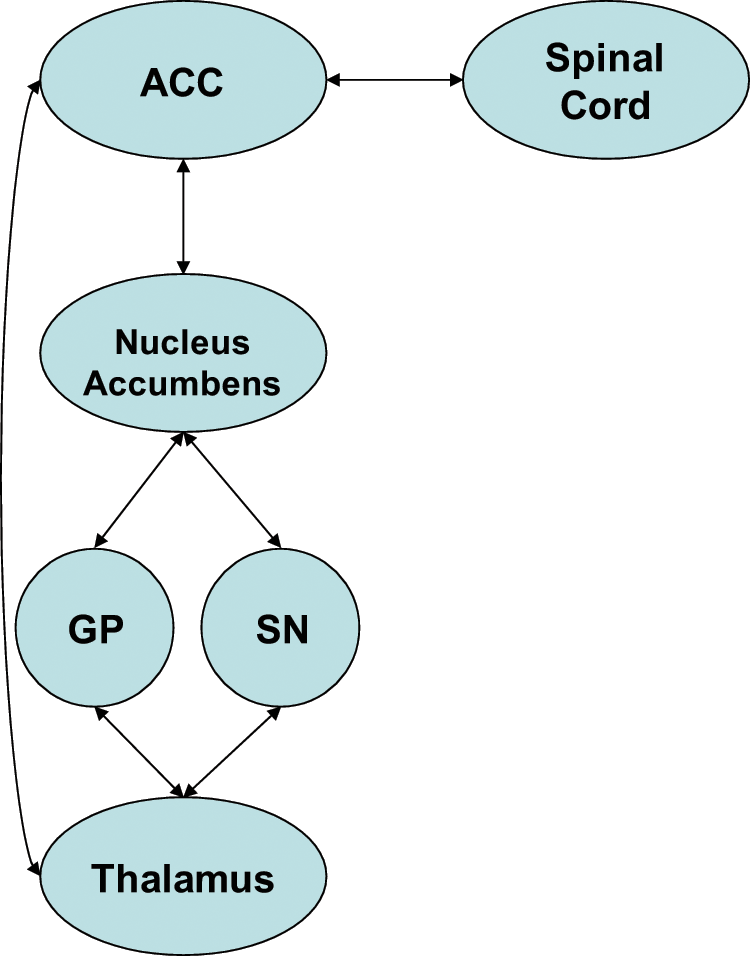
Anterior cingulate cortex circuit.
Cognitive and behavioural dysfunction mediated via frontostriatal circuits in FTD may be reflected in structural change in components of that circuitry (Looi et al., 2010). A meta-analysis of both structural and functional imaging of FTLD and controls demonstrated the existence of characteristic patterns of activation and atrophy for each of the subtypes of FTLD in: prefrontal cortex, temporal lobe, amygdala and neostriatum (Schroeter et al., 2007). Whilst FTLD has traditionally been considered to result from disease of the frontal or temporal cortex, the neostriatum itself may also be involved. The same neuropathological processes that affect the frontal and temporal cortex may also affect the neostriatum. There may be secondary neuroplastic changes due to trans-synaptic neurodegeneration arising from frontal or temporal pathology, or indeed pallido-thalamic pathology. Finally, the neostriatum has particular vulnerability to hypoxic, metabolic and vascular disease as a result of a limited vascular supply arising primarily from perforating terminal vessels from the circle of Willis.
The neostriatum serves as an entry point for afferent information from the periphery, as well as for afferents and efferents for functionally segregated regions of the cortex (Alexander et al., 1986; Haber, 2003; Haber et al., 2000). Grey matter volumes in intrinsic neural connectivity networks have been correlated with function in healthy controls (Seeley et al., 2009), and the neostriatum comprises a significant subcortical grey matter body. The corollary is that cortical atrophy may lead to loss of inputs to the neostriatum and neuroplastic reduction in the grey matter volume of the neostriatum. Similarly, there is evidence of deep white matter pathology between the frontal lobes and the neostriatum (Avants et al., 2010; Whitwell et al., 2010; Zhang et al., 2009), which may result in disconnection or diaschisis of frontostriatal circuits, with associated neuroplastic change in the neostriatum. It is possible that these white matter changes may arise from altered neuronal function in the white matter itself. Alternatively, the white matter disease may arise from secondary neuroplastic change due to cortical changes, and/or changes in other components of the circuits, such as the striatum, pallidum and thalamus.
Clinical syndromes of FTLD: Relationship to frontostriatal circuits
As they are presently constituted, the clinical subtypes of FTLD arguably cluster clinical features that may have a structural basis in the smaller-scale distributed neural networks of the frontostriatal circuits.
There have been attempts to differentiate the clinical subtypes of FTLD based upon neurobehavioural features and neuropsychological testing (Bozoki and Farooq, 2009). The clearest distinction is between FTD and the two language-disordered subtypes, PNFA and SD, in terms of behaviour. Clinically, FTD displays greater degrees of neurobehavioural disturbance, in the form of pathological behaviours, than either PNFA or SD (Bozoki and Farooq, 2009). That is, FTD patients display greater apathy, disinhibition, aberrant motor behaviour, eating disorder and agitation as assessed via the Neuropsychiatric Inventory (Bozoki and Farooq, 2009; Marra et al., 2007; Rankin et al., 2006; Rosen et al., 2006). Motivation is ascribed to a structural substrate in the anterior cingulate frontostriatal circuit (Cummings, 1993), thus the apathy seen in FTD may be due to dysfunction of this circuit. Similarly, maintenance of social comportment, and inhibition of disordered behaviours, has a structural basis in the orbitofrontal circuit; thus disinhibition, agitation and eating disorder may reflect dysfunction of this circuit (Cummings, 1993). Recently, there has been literature linking behavioural changes with their presumed substrates, such as the finding that right caudate/subcallosal gyrus grey matter density was correlated with empathy (Rankin et al., 2006). Aberrant motor behaviours in FTD may be associated with downstream dysfunction of the common putaminal component of a number of frontostriatal circuits (Josephs et al., 2008; Looi et al., 2010). Therefore, the neostriatum, as a component of frontostriatal circuits, may be a potential structural basis for FTD behavioural manifestations.
In addition, there is evidence of relatively more impairment on executive tasks in FTD compared to controls, and other neurodegenerative disease (Wittenberg et al., 2008). Such functions, such as planning, sequencing, verbal fluency, set-shifting and mental flexibility; are also served by primarily dorsolateral prefrontal frontostriatal circuits (Alexander et al., 1986; Cummings, 1993). In contrast, language impairments are more common in SD and PNFA, [soon to be reclassified as primary progressive aphasia, fluent and non-fluent (Wittenberg et al., 2008)], implicating temporal lobe structures and circuits other than frontostriatal.
However, there is evidence that, as PNFA progresses, the behavioural presentation ultimately resembles that of FTD (Banks and Weintraub, 2008; Bozoki and Farooq, 2009; Marczinski et al., 2004). The corollary is that, relative to SD, PNFA may manifest behavioural disorders secondary to frontostriatal circuit dysfunction involving the neostriatum, albeit to a lesser degree and of later onset than FTD. SD patients show lesser degrees of behavioural change, such as interruptive behaviour, perseveration, tangentiality and resisted redirection (Rankin et al., 2008).
Furthermore, a large longitudinal study of patients with several subtypes of FTLD showed persistent group differences on neuropsychological testing, with the caveat that there was a restricted range of tests included for the large sample (Libon et al., 2009). Allowing for the classification of the behavioural variant FTD by Libon et al. into an analogous SOC/EXEC impairment group, both the FTD and PNFA groups show a predominant executive dysfunction, compared to contrasting differences for SD on confrontational naming (Libon et al., 2009). Interestingly, this group states the executive dysfunction in PNFA may, in fact, be greater than in the FTD subtype.
The relevance of frontostriatal circuit or networks in the cognitive and behavioural presentation of FTLD has been increasingly recognised, and is the focus of ongoing investigation (Rabinovici et al., 2007; Wittenberg et al., 2008). Interestingly, homology of the neuropsychological profile of the behavioural variant FTD, the subtype with greatest executive dysfunction, has been found with Lewy body dementia, a neurodegenerative disorder significantly affecting the striatal system (Johns et al., 2009). As many of these pathways are routed through the neostriatum, this structure may provide a strategic view of the neuroanatomical basis of such presentations.
The neostriatum in FTLD: Neuropathological findings
Severe atrophy of the caudate and putamen was noted long ago in FTLD via post-mortem studies (von Braunmühl, 1930). Macroscopic neuropathology reveals that atrophy of basal ganglia is seen in a proportion of cases (Cairns et al., 2007), with the caveat that it may be difficult to detect mild striatal atrophy post-mortem (E. Englund, 2010, personal communication). Recent neuropathological studies have shown that early and specific neurodegeneration occurs in the striatum (Rabinovici et al., 2007; Whitwell et al., 2009a).
Overall, there is not a precise correspondence between the clinical syndromes and the identified molecular neuropathology in FTLD (Burrell and Hodges, 2010; Macedo et al., 2009). Macedo and colleagues (2009) have classified the molecular neuropathology corresponding to the overall macroscopic frontotemporal lobar presentation. Behavioural variant FTD has been associated with tau and TAR DNA-binding protein (TDP-43) molecular pathology. Of the tau-predominant pathologies, PNFA is often associated with clinical presentations of progressive supranuclear palsy (PSP) and corticobasal syndrome/degeneration (CBD). In SD, considered due to predominant left temporal atrophy, there may be tau, Pick bodies and Alzheimer’s disease pathology (up to 20%). Macedo et al. (2009) also describe a right temporal lobe variant with TDP-43, Pick bodies and Alzheimer’s disease pathology in up to 20%. For ease of reference we have reproduced a version of a table that summarises the most recent molecular neuropathological findings for the major FTLD subtypes: FTD, SD and PNFA (Burrell and Hodges, 2010; Rabinovici and Miller, 2010) (Table 1).
FTLD subtype by molecular pathology
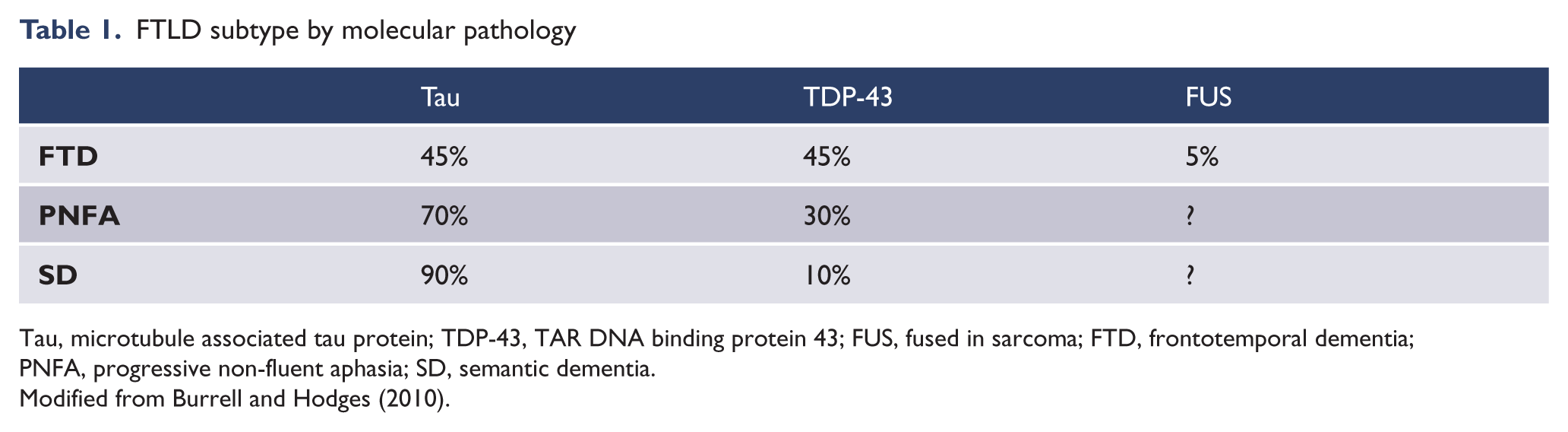
Tau, microtubule associated tau protein; TDP-43, TAR DNA binding protein 43; FUS, fused in sarcoma; FTD, frontotemporal dementia; PNFA, progressive non-fluent aphasia; SD, semantic dementia.
Modified from Burrell and Hodges (2010).
The most common molecular pathological changes in FTLD are tau-negative, ubiquitin-immunoreactive intraneuronal inclusions that frequently stain positive for TDP-43 and are often associated with mutations in the progranulin (PGRN) gene (FTLD-TDP). FTLD-TDP may preferentially affect cortical regions and hippocampus, but also shows striatal atrophy.
The next most common neuropathological change is FTLD-tau, known as Pick’s disease. Pick bodies are found in neocortex, subiculum, hippocampus, entorhinal cortex and subcortical (including striatal) regions (Neumann et al., 2009). In the other tauopathy-associated subtypes of PNFA, progressive supranuclear palsy and corticobasal degeneration, tau accumulation is prominent in striatal regions (Cairns et al., 2007).
Previously identified as the ubiquitin-positive, tau- negative FTLD, FTLD-FUS (fused in sarcoma) is associated with amyotrophic lateral sclerosis (ALS) and shows marked striatal neurite damage and atrophy (Mackenzie and Rademakers, 2007; Mackenzie et al., 2006; Neumann et al., 2009). An earlier age of onset and more rapid decline has been found in FTLD-FUS, as has marked striatal atrophy (Davion et al., 2007; Leverenz et al., 2007), and is associated with asymmetric motor disturbance (Yokota et al., 2009). Extreme atrophy of the caudate has been observed at autopsy and ante-mortem in FTLD-FUS (Josephs et al., 2010).
Frontotemporal dementia with motor neuron disease (FTD-MND) also shows marked striatal atrophy (Seelaar et al., 2007). On VBM of MRI, patients with mutations in the microtubule-associated protein tau (MAPT) showed greater putaminal atrophy than those with the PGRN mutation (Whitwell et al., 2009b). In FTD and Parkinsonism linked to chromosome 17 associated with the tau gene mutations (FTDP-17T), macroscopic atrophy is detectable at the intermediate stage in the caudate nucleus, whilst caudate and putaminal atrophy may be more evident in advanced illness (Ghetti et al., 2003).
Thus, aside from the cortical neuropathology associated with the molecular subtypes of FTLD, there is convergent evidence of involvement of the striatum, which may be as a result of the particular strategic susceptibility of this structure to metabolic, hypoxic or vascular disease (I. Mackenzie, 2010, personal communication). This sensitivity of the striatum as a potential marker is operationalised in the consensus algorithms for neuropathological diagnosis of FTLD, which involve staining and investigation of the striatum (Cairns et al., 2007).
There has been evidence that other neuropathological processes may affect the neostriatum, such as beta-amyloid deposition in Alzheimer’s disease (Villemagne et al., 2009), and accordingly there may be considerable overlap in the accumulation of neurodegenerative burden. This may be illustrated in behavioural phenocopy conditions such as progressive logopenic aphasia, which clinically presents with features in common with SD/PNFA, but which arises primarily from AD pathology (Gorno-Tempini et al., 2008). Beta-amyloid deposition has also been described in clinical FTD cases, confirming the possibility of overlap in molecular pathology (Hu et al., 2011). The specific vulnerability of the morphology of the neostriatum to parallel disease processes may be a sensitive marker of disease, correlation of which to clinical features and molecular pathology may yield insights into the relative contributions of various neurodegenerative processes.
Neurochemical CSF studies of FTLD
In vivo CSF studies have focused on the role of biomarker analysis, and some studies have identified that FTLD subtypes differ from AD and each other in CSF biomarkers (Hu et al., 2010), although there remain a profusion of candidate biochemical markers, the relative roles of which remain uncertain (Hu et al., 2011). AD may be distinguished from FTLD by p-tau181 (rather than total tau), Aβ42, and agouti-related peptide (AgRP); whilst the FTLD-tau neuropathological subtype may potentially be distinguished from FTLD-TDP by adrenocorticotropic hormone (ACTH), eotaxin-3, Fas, and interleukin 17 (IL-17) (Hu et al., 2010). Neither Aβ42 or, surprisingly, CSF tau differed between FTLD-tau or FTD-TDP (Hu et al., 2010). At this stage, these CSF biomarkers require further validation, although their presence, combined with the neuropathological findings discussed above, supports the view that there may be a convergence of neurodegenerative processes in FTLD, some of which may localise to the neostriatum. There is a potential role for multiple biomarker cross-validation, including: genotype, molecular pathology, neuropathology, CSF analytes, neuroimaging, gross neuroanatomy and clinical features; through the striatum.
Neuroimaging of the neostriatum in FTD
Our recent neuroimaging research (Looi et al., 2008, 2009) and that of others (Chow et al., 2008; Garibotto et al., 2011) has identified that subcortical atrophy in the neostriatum (caudate nucleus and putamen) may be a crucial factor underlying the cognitive, emotional and behavioural decline of FTD in vivo. Furthermore, this striatal atrophy has been demonstrated using methodologically distinct MRI analysis methods: manual segmentation (Looi et al., 2008, 2009), automated parcellation (Chow et al., 2008) and probabilistic mapping (Garibotto et al., 2011). These in vivo neuroimaging findings converge with recent neuropathological studies demonstrating that specific intraneuronal inclusions and atrophy are present in the neostriatum, especially in younger-onset familial forms of FTLD (Davion et al., 2007; Leverenz et al., 2007; Seelaar et al., 2007), in autopsy-confirmed FTLD (Rabinovici et al., 2007, Whitwell et al., 2009a); and with evidence of associated circuit atrophy, present in early clinically diagnosed FTD (Seeley et al., 2008).
Recently, correlations between cortical thickness and white matter integrity measured using diffusion tensor imaging have been found in FTLD, demonstrating patterns distinguishing AD and FTLD, with greater white matter disease in the genu of the corpus callosum, left uncinate, left inferior fronto-occipital fasciciulus, cingulum and bilateral corona radiata being linked to cortical changes in anterior cingulate, middle, inferior and orbitofrontal gyri (Avants et al., 2010). Of these structures, the cingulate and frontal gyri of the cortex, as well as the cingulum and corona radiata, may have interconnections to the neostriatum. Through either secondary neuroplastic (trans-synaptic neurodegeneration) or shared neurodegenerative processes the neostriatum may also be affected. Similarly, there has been evidence of increased mean diffusivity, and hence reduced structural integrity of grey matter in frontal and temporal regions in FTLD, associated with mean diffusivity changes in white matter tracts to the frontal lobes (Whitwell et al., 2010). Again, this implies a possible role for altered structural integrity of white and grey matter in FTLD impacting upon the neostriatum.
Therefore, striatal atrophy may be an early and sensitive biomarker for FTD, especially in the younger-onset familial forms of FTD. Similarly, since early specific neuropathology is present in the neostriatum in FTD, there is potential that neostriatal atrophy may be a biomarker for preclinical FTD, as has been proposed in Huntington’s disease (Aylward, 2007). Thus, there is convergent evidence that structural change may arise in the neostriatum due to both neuronal level molecular pathological neurodegeneration and frontostriatal circuit nodal morphological change in white or grey matter. As these findings converge upon the neostriatum, so the neostriatum may prove to be a sentinel marker for early FTD.
Neostriatal atrophy in subtypes of FTLD
We hypothesised that frontostriatal neurodegeneration may be the structural basis of clinical manifestations of FTLD, and found that significant neostriatal atrophy was evident in FTLD, but not in controls or Alzheimer’s disease (Looi et al., 2008, 2009). Independently, Rabinovici et al. (2007) concluded that, in their autopsy-confirmed group with FTLD, there was evidence of degeneration in the frontostriatal network in FTLD in comparison with Alzheimer’s disease, with specific atrophy in the anterior cingulate, frontal insula, subcallosal gyrus and striatum in FTLD.
There is also converging evidence that the behavioural variant FTD is associated with greater neostriatal atrophy than other subtypes of FTLD, as might be expected based upon the observation the clinical phenotype of FTD is essentially that of frontostriatal dysfunction. Our study showed there was a gradient in severity of atrophy, such that FTD showed more atrophy than PNFA and SD (Looi et al., 2008). Chow et al. (2008) demonstrated specific striatal atrophy in FTD, whilst Garibotto et al. (2011) demonstrated bilateral striatal atrophy in FTD. Using VBM, Seeley et al. (2009) demonstrated syndrome-specific regional atrophy in FTLD subtypes, finding specific atrophy in the striatum in FTD, and our interpretation of their atrophy patterns suggests involvement of striatum in both SD and PNFA. Also using VBM, Whitwell et al. (2009b,c) studied ‘subtypes’ of FTD, classifying into four groups based on the relative lobar atrophy: temporal dominant, temporo-fronto-parietal, frontotemporal and frontal dominant. Both frontotemporal and frontal dominant subgroups of FTD showed atrophy of the caudate nucleus, and in the putamen in the frontotemporal group. Thus, there is evidence that neostriatal degeneration may be both specific to FTLD in comparison with Alzheimer’s disease, and be more prominent in subtypes with greater frontostriatal dysfunction, such as FTD, rather than PNFA or SD.
Towards a frontostriatal endophenotype for FTD
The concept of an endophenotype has been employed to investigate neuropsychiatric disease, such as FTLD: that is, a restricted set of phenotypic or clinical features that may have a more specific structural and hence, genetic basis (Weiser et al., 2005). An example of an endophenotype is frontal-executive neuropsychological function, localised to the neural substrate of dorsolateral prefrontal cortex frontostriatal circuit, which has been investigated fruitfully in schizophrenia. Consequently, it is possible to explore the structural basis of an endophenotype by studying the components of neural circuits carrying such functions. As the frontostriatal circuits that traverse the neostriatum serve specific cognitive, emotional and behavioural functions which can be assessed via neuropsychological testing, clinical scales and observations, the combination of the neostriatal biomarker and clinical features constitutes a potential frontostriatal endophenotype.
Regional morphological change, in addition to volumetric change, in the neostriatum may allow us to quantify the neurostructural basis of the frontostriatal endophenotype in FTD. Neostriatal components are ideal candidate structures for morphological or shape analysis due to the highly specific nature of their regional interconnections. Thus, morphological analysis (Thompson, 1945) may be a useful tool to quantify neurodegenerative atrophy, manifest as shape change in FTD.
Morphology of the neostriatum as a map of frontostriatal circuit neurodegeneration
The caudate and putamen are divided into highly specific functional subregions based on afferents received from the frontal cortex and substantia nigra (Haber, 2003; Haber et al., 2000). The caudate head and body receives connections on its lateral aspect from the dorsolateral prefrontal cortex, inferior orbitofrontal cortex, and posterior parietal cortex, whereas the tail receives input from the frontal eye fields. On its medial aspect, the caudate is connected to the anterior cingulate cortex (Alexander et al., 1986; Haber et al., 2000; Heimer and Van Hoesen, 2006; Utter and Basso, 2008). The putamen receives connections on its medial aspect from the motor cortex, and somatosensory cortex. The lateral putamen receives connections from the supplementary motor area. Haber et al. (2000) and Haber (2003) also described connections from the dorsolateral prefrontal cortex to the ventral aspect of the putamen. In addition, there are fibres traversing the internal capsule linking the caudate and putamen above their mutual origin in the nucleus accumbens (Alexander et al., 1986; Draganski et al., 2008; Heimer and Van Hoesen, 2006; Utter and Basso, 2008). Regional changes in the shape of the caudate and putamen may thus be associated with dysfunction of specific circuits with resultant clinical consequences (Figures 5 and 6).
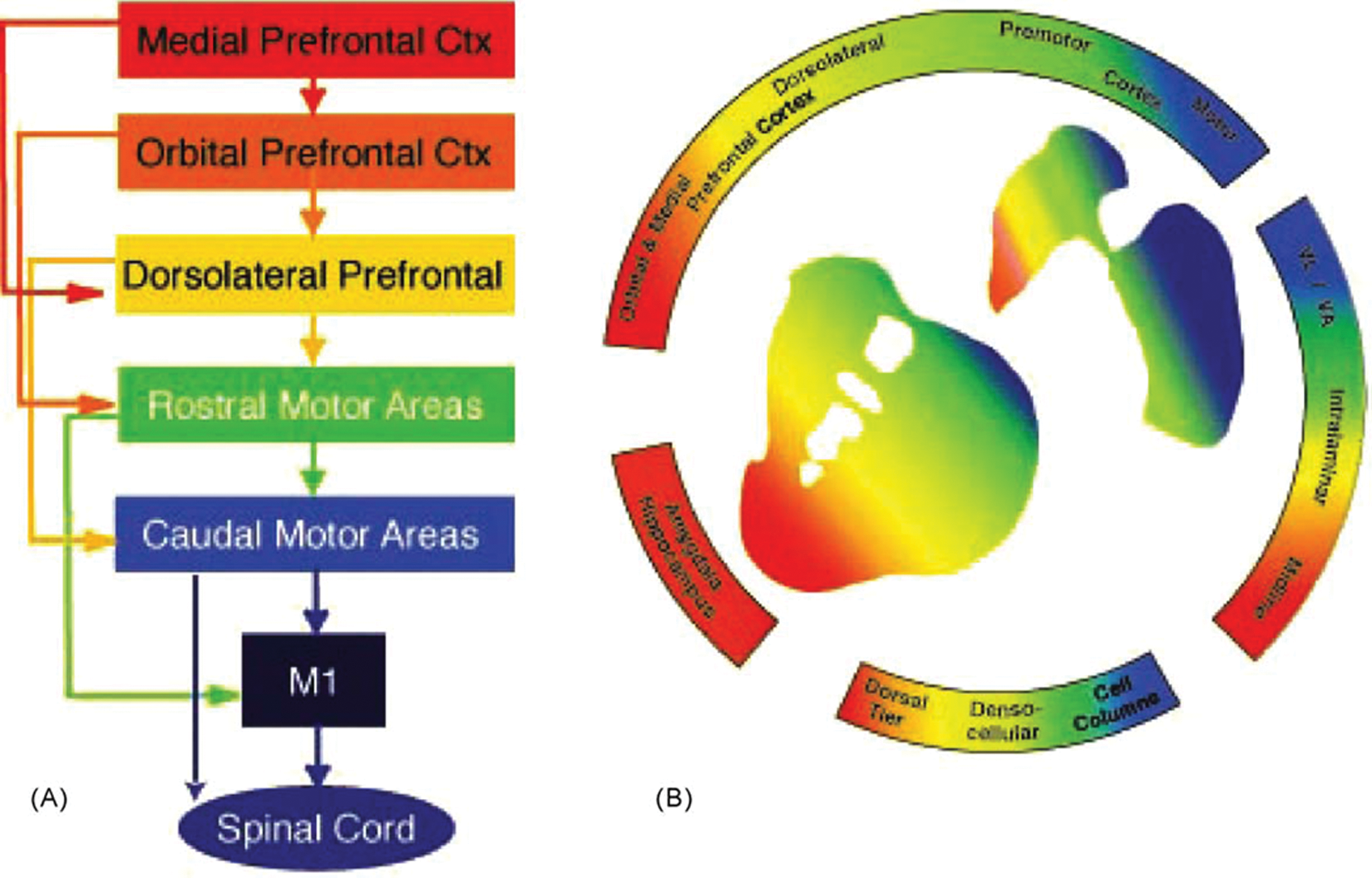
Diagram demonstrating the functional organisation of the frontal cortex and striatal afferent projections. (A) Schematic illustration of the functional connections linking frontal cortical brain regions. (B) Organisation of cortical and subcortical inputs to the striatum. In both (A) and (B), the colours denote functional distinctions. Blue: motor cortex, execution of motor actions; green: pre-motor cortex, planning of movements; yellow: dorsal and lateral prefrontal cortex, cognitive and executive functions; orange: orbital prefrontal cortex, goal-directed behaviours and motivation; red: medial prefrontal cortex, goal-directed behaviours and emotional processing.
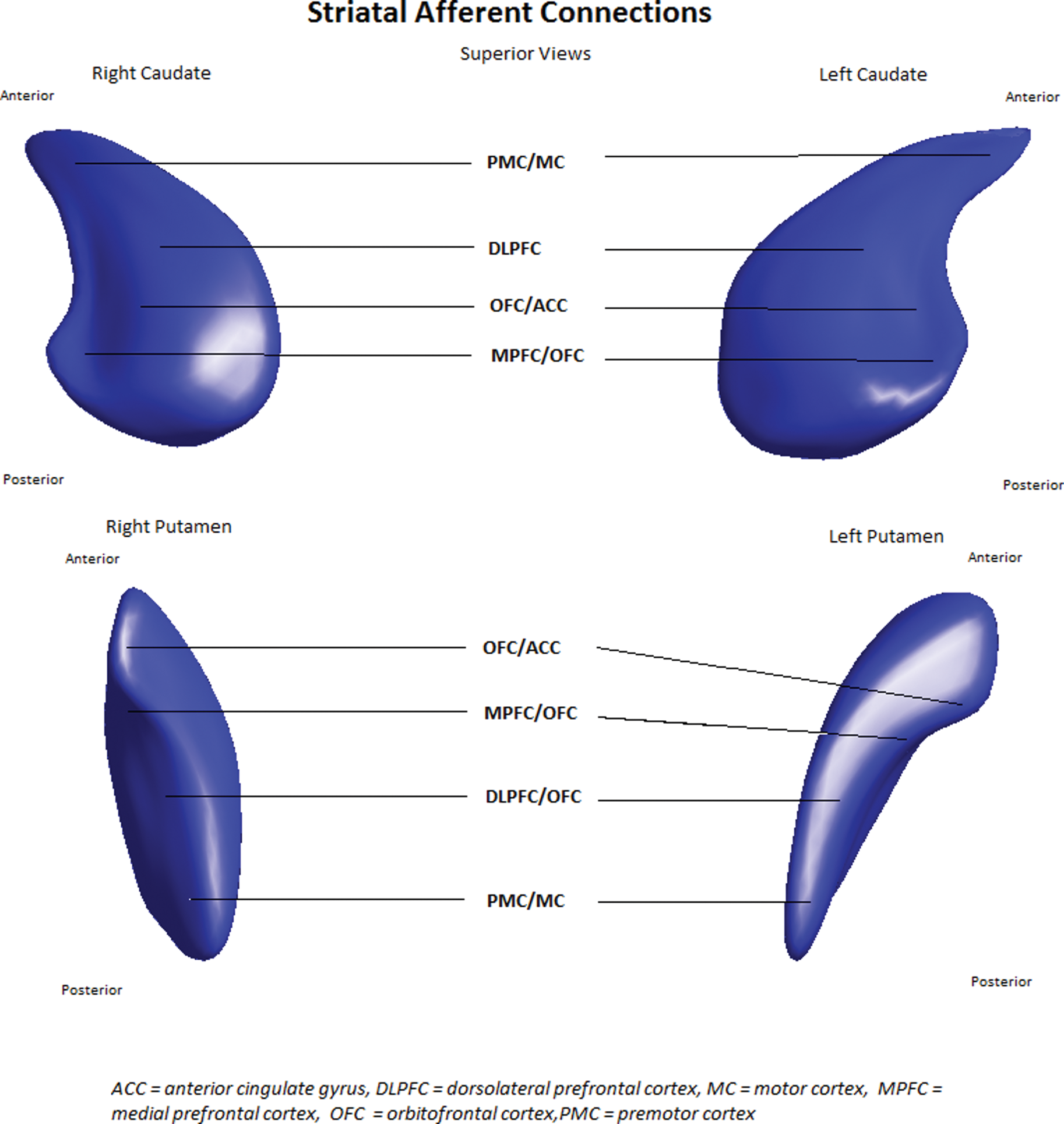
Map of striatal afferents.
Spherical harmonic shape analysis (SPHARM) of the caudate and putamen is able to resolve localised areas of morphological change. Levitt et al. (2009), Styner et al. (2006), Hwang et al. (2006), Choi et al. (2007) and Mamah et al. (2008) have previously applied shape analysis to the striatum in schizotypal personality disorder, bipolar disorder, obsessive–compulsive disorder and siblings of persons with schizophrenia, respectively. Levitt et al. (2009) found an association between shape deflation and verbal learning capacity in females with schizotypal personality disorder, implicating frontostriatal connections to the ventromedial prefrontal cortex. We have applied shape analysis to the congenital genetic disorder choreoacanthocytosis (ChAc) with significant findings of neostriatal shape and volume change compared to controls (Walterfang et al., 2011). As ChAc is a specific genetic disorder, resulting in isolated atrophy of the neostriatum, this supports the case for frontostriatal circuits as a potential structural basis for the endophenotype of obsessive–compulsive phenomena and frontal-executive dysfunction seen in ChAc. Furthermore, we found altered striatal morphology in progressive supranuclear palsy, a Parkinson’s plus syndrome in which abnormal tau accumulates in the striatum, in association with a dysexecutive syndrome, apathy and other frontostriatal system clinical features (Looi et al., 2011b). Similarly, the frontostriatal endophenotype is relevant for FTLD.
Using SPHARM, we investigated the shape of the neostriatum as a map of regional atrophy in relevant frontostriatal circuits in FTLD, AD and controls (Looi et al., 2010, 2011a, finding that there are regionally specific shape deflations in the neostriatum, and thus potential neural substrates for a frontostriatal endophenotype in FTLD. We have also found that the neostriatal shape and volume differences in subtypes of FTLD such as behavioural variant FTD, SD and PNFA correspond with the expected degree of frontostriatal dysfunction, with greatest atrophy seen in FTD, followed by PNFA and SD (Looi et al., 2008, 2009). Therefore, the neostriatum may also be a biomarker useful for subtyping or, more specifically, endophenotyping of FTLD.
Patterns of morphological change in FTLD and neurodegeneration
In our shape analyses of FTLD and subtypes, we found there was significant shape and volume difference between 34 patients with FTLD, 19 patients with Alzheimer’s disease (AD) and 27 controls (Looi et al., 2010, 2011a). When all FTLD subtypes were combined, there was significant shape deflation in the left caudate compared to AD, and bilaterally compared to controls, with significant but lesser changes in the putamen (Looi et al., 2010). When we investigated the FTLD subtypes, we found that the FTD subtype appeared to be the main contributor to the shape deflation in the caudate, followed by PNFA and minimal change in the SD subgroup; and again, lesser changes in the putamen, compared to controls (Looi et al., 2011a).
Our shape analysis identified both general and specific patterns of shape deflation with FTLD (Looi et al., 2010, 2011a). The pattern of shape deflation may correspond to distance from sources of neurogenesis for the caudate. There are at least two regions adjacent to or within the caudate in which neurogenesis may occur: the shell of the nucleus accumbens (Heimer and Van Hoesen, 2006), and the subventricular zone comprising the medial border of the caudate (Curtis et al., 2007). We hypothesised that distance from neurogenic zones may influence the rate and extent of reactive neurogenesis in response to neurodegeneration, analogous to the pattern of morphogenesis observed with chemical gradients in living organisms (Thompson, 1945; Turing, 1952). That is, the more distant the surface of the structure from a neurogenic source, the less likely it is to receive new neurons, and thus the more likely it is to show atrophy, manifest as shape deflation in a gradient notionally inversely proportional to the distance from the source. We observed two gradients of atrophy in the caudate in FTLD consistent with the distance from a neurogenic origin in the nucleus accumbens: dorsal–ventral and posterior–anterior. A dorsal–ventral gradient of atrophy has also been described in other neurodegenerative disease affecting the caudate and putamen: Huntington’s disease (Douaud et al., 2006) and choreoacanthocytosis (Walterfang et al., 2011). We also observed a lateral–medial gradient of atrophy, corresponding to distance from the subventricular zone. Thus, our shape analysis identified three patterns of shape deflation, which may arise from general patterns of neurodegeneration affecting the neostriatum.
In FTLD, we also found specific regional atrophy of potential functional significance, affecting the interconnections of the neostriatum to dorsolateral prefrontal, anterior cingulate and orbitofrontal cortex; motor cortex corresponding to the frontal eye fields in the caudate; and primary motor cortex in the putamen (Looi et al., 2010). Thus, based upon the findings of general and specific patterns of neostriatal morphological change in FTLD, we suggest correlative neuropathological studies should be conducted to determine the microscopic/molecular bases of these patterns.
Our most recent shape analysis of the clinically diagnosed subtypes of FTLD demonstrated that the majority of the neostriatal atrophy and thus, morphological change, could be attributed to changes in the FTD subtype, and to a lesser extent the PNFA subgroup, in comparison to controls (Looi et al., 2011a). Again, the shape deflation was most pronounced in the FTD subtype of FTLD, the subtype with the greatest putative frontostriatal dysfunction. The FTD subtype regional atrophy was of functional significance, affecting the regions believed to be interconnected to the frontostriatal circuits. Notably, there was no significant neostriatal shape difference of the SD subtype in comparison with controls, such that neostriatal shape change appeared in the gradient FTD > PNFA >SD ≥ controls. We note, however, the shape analysis was not sufficiently powered to investigate neostriatal shape differences between subtypes of FTLD.
Given that the frontostriatal white matter connects the neostriatum with the neocortex, it is possible that the specific regional atrophy in the neostriatum may also be associated with atrophy in frontostriatal white matter and the frontal cortex (Avants et al., 2010; Matsuo et al., 2008; Whitwell et al., 2010; Zhang et al., 2009). Similarly, such a relationship may also exist for corticostriatal circuits. The mechanism for these disconnection phenomena may include diaschisis and trans-synaptic degeneration, the process by which anterograde and retrograde neuronal effects arise from neuronal damage (Buren, 1963). The morphology of the white matter may be assessed by diffusion tensor MRI, whilst cortex morphology may also be quantified. Potentially, then, in addition to the morphology of the neostriatum as a strategic biomarker, the morphology of frontostriatal white matter and the frontal cortex may also serve as biomarkers, linked to clinical features and stage of FTLD.
Conclusions and future directions
We propose the investigation of the FTD subtype of FTLD as a frontostriatal disorder, through morphological measurement of significant structural components such as the neostriatum, frontostriatal white matter tracts, the frontal cortex and the functions served by these circuits. The neostriatum and connections may prove to be valuable early biomarkers in FTD: for early diagnosis; staging illness; subtyping or endophenotyping based upon the pattern of atrophy; and ultimately, the design of targeted treatments. In relation to subtypes of FTLD without significant neostriatal atrophy, such as SD and to a lesser extent, PNFA, it will be important to explore other circuit dysfunction, such as that of language circuits, as a potential endophenotype. Accordingly, frontostriatal features are not hypothesised as being universal to all subtypes of FTLD. However, frontostriatal disorder may account for at least some features of FTD and PNFA, if not SD. Thus, our proposal converges at the super-ordinate level with the investigation of the selective vulnerability of large-scale human brain networks (Seeley et al., 2009), and will contribute to understanding of frontostriatal networks as a subset of larger networks. Practically, we have demonstrated methods for quantifying the morphology of the strategically central neostriatum, which can thus serve as a window into the structural, and hence functional integrity, of frontostriatal circuits (Looi et al., 2010, 2011a). In future, it may also be possible to correlate the structural morphology of frontostriatal circuits with neuropsychological functions and measurements of neuropsychiatric features (depression, apathy, mania) carried by those circuits.
An understanding of the pathophysiology of FTD based on the neostriatum may yield proximate and future insights into treatment strategies. At the proximate level, the quantification of the degree of frontostriatal circuit dysfunction via correlation of the neuroimaging and clinical biomarkers may inform the design of psychological and occupational therapy interventions. In the future, based upon correlation with genotype, molecular pathology and CSF biomarkers, drug treatment targets for abnormal pathophysiology may be identified, the success of treatment of which may be measured by quantification of neostriatal morphology using such methods as we have shown. Highly speculative, but possible based on stem cell treatment in Parkinson disease and other neurodegenerative disease (Lunn et al., 2011), is the potential targeted promotion of neurogenesis in the subventricular zone (Curtis et al., 2007) adjacent to the striatum, and the core of the nucleus accumbens (Heimer and Van Hoesen, 2006) within the striatum; informed by understanding of the patterns of neurodegeneration via the methods we espouse.
In re-conceptualising FTD as a frontostriatal disorder, we believe that in vivo morphology via MRI may serve as a useful interconnection between molecular neuropathology and clinical features; that is, as a useful endophenotype, an approach similar to that adopted in advancing understanding of neuropsychiatric disease (such as in choreoacanthocytosis, bipolar disorder and schizophrenia). In addition, such study will also contribute to the understanding of large-scale human brain networks, of which the frontostriatal circuits serve as a sub-system. By understanding neural morphology, manifest in the warp and weave of the fabric of the frontostriatal circuits, we may discern the patterns of disease in the tragic tapestry of FTLD.
Footnotes
Author contributions
JCLL conceived, designed and is the guarantor of the FTLD volumetric and shape analysis studies on which the paper has been based and wrote the first draft of this paper. MW (principal shape analyst) and DV (collaborator on shape analysis) were principal investigators on the shape analysis of FTLD on which the paper has been based. LS (software for image pre-processing and analysis) and L-OW (Karolinska University Hospital Huddinge FTLD cohort) were principal investigators of the FTLD studies on which the paper has been based. MDM assisted with writing the paper and produced ![]() . All authors contributed to writing and editing of the paper.
. All authors contributed to writing and editing of the paper.
Acknowledgements
Research reported here made use of the SMILE medical imaging laboratory at Karolinska University Hospital, Stockholm, Sweden. JCLL was supported by the Canberra Hospital Specialists Private Practice Trust Fund, ACT health funding, and self-funding for travel and accommodation for research conducted at SMILE and Melbourne Neuropsychiatry Centre. The contributions of the volunteers and collaborators of the morphological studies of FTLD conducted at SMILE are also gratefully acknowledged. Collaborators who contributed to the papers on which this review is, in part, based include: Christian Andersen, Lisa Botes, Phyllis Chua, Eva-Lena Engman, Rajeev Kumar, Olof Lindberg, Eva Örndahl, Per Östberg, Martin Styner, Marc Niethammer, and Bram B. Zandbelt.