
Abstract
Background
Hypophosphatasia is a rare inherited metabolic disease resulted by ALPL gene mutations. It is characterized by defective bone and teeth mineralization. The phenotypic spectrum is highly variable ranging from lethal perinatal form to mild forms which are only diagnosed in adulthood or remain undiagnosed despite persistently low concentrations of ALP. The aim of this study is to evaluate the clinical phenotype and frequency of ALPL mutations in a group of patient with hypophosphatasaemia.
Methods
Thirty individuals with alkaline phosphatase values below 40 IU/L in at least two assessments and having no alternative explanation for their low ALP concentrations were included in the study. The clinical features and radiological data of the study group were re-investigated for hypophosphatasia-related findings. ALPL sequence analysis was performed using Sanger sequencing.
Results
No patient in the study group had severe symptoms, nor had they initially been diagnosed as having hypophosphatasia. Four different heterozygous ALPL mutations (c.542C>T, c.648 + 1G>A, c.657G>T and c.862 + 1G>C) were found in four patients. One splice site mutation (c.862 + 1G>C) was reported for the first time in this study.
Conclusion
ALPL sequence analysis may help to diagnosing genetic defects in individuals with persistently low ALP concentrations and provide to take preventive measures before symptoms appear. As in the other populations, HPP displays allelic heterogeneity in our population.
Introduction
Hypophosphatasaemia, a low serum alkaline phosphatase (ALP) concentrations, is associated with a broad and variable clinical spectrum ranging from lethal neonatal skeletal form to odonto hypophosphatasia (HPP) or arthritic problems in adult life.1,2 In most cases, low ALP is accompanied by various unrelated systemic disorders and may be incidentally discovered. 3 It is caused by loss of function mutations in the gene (ALPL) which encodes a tissue-nonspecific isoenzyme of alkaline phosphatase (TNSALP). The main function of TNSALP is to cleave phosphocompounds such as inorganic pyrophosphate (PPi), pyridoxal-5-phosphate (PLP) and phosphoethanolamine (PEA). 4 The functional defects of TNSALP lead to the extracellular accumulation of these phosphate compounds, thus causing dental and bone mineralization defects, in conjunction with other clinical findings. 5 PPi accumulation inhibits the synthesis of hydroxyapatite crystals and growth – the main mechanism underlying the cardinal clinical features of HPP. PLP is the phosphorylated form of pyridoxine (vitamin B6). TNSALP dephosphorylates PLP. The dephosphorylation of PLP by TNSALP is a necessary step for crossing blood–brain barrier and neuronal uptake. Pridoxal is then phosphorylated to PLP into the cells, where it plays a role as a co-factor in gamma-aminobutyric acid synthesis. Therefore, in HPP, inability to cleave PLP may lead to vitamin B6 deficiency in central nervous system and seizures.4,6
ALPL gene is located on chromosome 1p36.12 and consists of 12 exons. The first and initial part of the second exon is non-coding. Its product is a protein of 524 amino acids.1,4 In the ALPL gene, around 400 mutations have been reported to date. 7 The majority of them are missense and the inheritance pattern can be either autosomal recessive (AR) or dominant (AD). Severe HPP phenotypes are usually associated with AR inheritance, whereas AD or AR inheritance can be found in mild HPP. In a number of ALPL mutation carriers with persistently low levels of ALP do not show overt clinical manifestations. However, it has been shown that subclinical abnormalities of bone remodelling can be present in those individuals. A recommendation to avoid antiresorptives such as bisphosphonates should be given in those cases. 8 Therefore, early diagnosis is important in such individuals.
Clinical heterogeneity makes the diagnosis of HPP challenging. Because of difficulties in the diagnosis of asymptomatic HPP carriers or late onset adult types, persistently low ALP activities in the paediatric age group can be considered a warning signal requiring further diagnostic work-up.
In this study, we aimed to investigate the proportion of ALPL mutation carriers among children and adolescents with low ALP concentrations in conjunction with phenotypic features of hypophosphataemic individuals with ALPL mutations.
Materials and methods
Patients
All patients aged between 0 and 23 years with alkaline phosphatase concentrations measured in the Clinical Biochemistry Laboratory, Ege University Faculty of Medicine (in Izmir/Turkey) between January 2014 and September 2018 were screened for their ALP values. After reviewing their clinical, radiological and laboratory records, 126 individuals with alkaline phosphatase values below 40 U/L in at least two assessments and having no alternative explanation (electrolyte abnormalities, drug use and hypothyroidism) for ALP depletion were the suitable candidates to the study from this group. A telephone call was placed to the 126 individuals for determining their interest in our study. Thirty subjects (3 males and 27 females) aged between 6 and 22.5 years old agreed to participate in the study. Informed consent was obtained from all individual participants included in the study. In the case of subjects under the age of 18 years, informed consent was signed by a parent and/or legal guardian. All individuals in the study group were clinically and radiologically re-investigated for their HPP-related symptoms and clinical findings. In patients found to have ALPL mutations, prospectively, serum total ALP, Ca, P and urine Ca concentrations measurements, radiography studies and bone mineral density measurements were performed.
Samples from the patients were obtained in accordance with the Helsinki Declarations. Written informed consent for genetic testing was obtained from all cases or their parents/guardians.
Biochemical analyses
ALP
Serum ALP was measured by colorimetric assay in accordance with a standardized method. In the presence of magnesium and zinc ions, p-nitrophenyl phosphate is cleaved by phosphatases into phosphate and p-nitrophenol. The p-nitrophenol released is directly proportional to the catalytic ALP activity. This is determined by measuring the increase in absorbance. The lower detection limit of ALP is 5 IU/L with a measuring range of 5–1200 U/L. ALP assay follows the recommendations of the IFCC. Intermediate precision (CV%) determined using control sera was 2.4% (Cobas Alkaline Phosphatase acc. to IFCC Gen2, Roche Diagnostics GmbH, Mannheim).
In our hospital, the ALP reference range lower limit is 40 U/L at the age of >18 years, regardless of gender. The reason why we chose the cut-off value of 40 U/L was to be able to screen all children and adolescents whose ALP activity was at the lowest reference limit of the adults’ range and much lower than the paediatric ages. By choosing a strict cut-off value rather than the lowest reference limit for paediatric ages, the sensitivity and the specificity were increased.
Calcium
Serum/urine Ca was measured photometrically. Serum Ca ions react with 5-nitro-5′-methyl-BAPTA under alkaline conditions to form a complex, which reacts with EDTA. The change in absorbance is directly proportional to the calcium concentration. The measuring range of serum Ca is 0.20–5.0 mmol/L (0.8–20.1 mg/dL) and urine Ca is 0.20–7.5 mmol/L (0.8–30.1 mg/dL). Urine Ca results have been given as a Ca/creatinine ratio. Intermediate precision (CV%) determined using control serum samples was 0.8% and control urine was 1.5% (Cobas Ca Gen2, Roche Diagnostics GmbH, Mannheim).
Phosphate
Serum phosphate was measured based on the reaction of phosphate with ammonium molybdate to form ammonium phosphomolybdate complex without reduction. The complex is determined photometrically in the (340 nm) ultraviolet region. The measuring range of serum phosphate is 0.1–6.46 mmol/L (0.3–20 mg/dL). Intermediate precision (CV%), determined using control serum samples, was 1.2%. (Cobas Inorganic Phosphorus Gen2, Roche Diagnostics GmbH, Mannheim).
All chemistry parameters were measured on Roche, Cobas® 8000 modular analyser (Roche Diagnostics).
Sequence analysis of ALPL
Genomic DNA of the probands was extracted from peripheral blood leukocytes using the QIAamp DNA Blood Kit (Qiagen, Germany). All coding regions and exon–intron boundaries of the ALPL were amplified by PCR using the primers given in the Table 1. The PCR products were sequenced using the BigDye Terminator v3.1 Cycle Sequencing Kit and migrated on capillary 3130 Genetic Genetic Analyser (Applied Biosystems, Foster City, CA, USA). Sequence alignment was performed using the CLC Genomics Workbench. 9 Pathogenicity of the variants was subsequently evaluated using several in silico tools: SIFT, MutationTaster, REVEL and CADD.10–13 Additionally, we applied the ACMG guidelines (2015) to classify the variants according their criteria. 14 The frequencies of the putative pathogenic variants are investigated in different databases: NCBI dbSNP build141 (http://www.ncbi.nlm.nih.gov/SNP/), 1000 Genomes Project (http://www.1000genomes.org/), Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org/) and Genome Aggregation Database (gnomAD) (http://gnomad.broadinstitute.org/). We additionally searched the ALPL gene mutation database (http://www.sesep.uvsq.fr/03_hypo_mutations.php) 7 as well as the Human Gene Mutation Database (HGMD) (http://www.hgmd.cf.ac.uk/ac/search.php).
Primer set for PCR amplification of ALPL gene.

Radiography and bone mineral density
Radiography studies were performed at the Department of Radiology, Faculty of Medicine Ege University. Skull and chest X-rays, and bilateral upper and lower extremity radiographs were taken using conventional X-ray radiography. The bone mineral density (BMD) was measured using dual energy X-ray absorptiometry densitometer (DXA, GE Lunar, USA) at Department of Nuclear Medicine. Height and weight of the subjects were measured using conventional equipment.
Results
From 30 patients having ALP concentrations lower than 40 IU/L (measured at least twice), four patients were found to be heterozygous for ALPL mutation. Four different mutations were identified in those patients (Table 2). One of four, c.862 + 1G>C, was novel, whereas other three were known mutations (c.542C>T, c.648 + 1G>A, c.657G>T). However, neither of these four mutation has been described in Turkish population so far. No patient in the study group had severe HPP symptoms, nor had they initially been diagnosed as having HPP. On re-examination with their medical histories reanalysed, it was observed that four patients (two of mutation-positive patients and two of mutation-negative patients) had experienced bone fractures. Two mutation-negative patients and one mutation-positive patient had short stature. Dental abnormalities were observed in two mutation-positive and two mutation-negative patients. Skeletal deformities (scoliosis in Patient 3 and genu valgus in Patient 4) were detected in two of the mutation-positive patients. Serum Ca, P and urine Ca and creatinine concentrations were found to be normal in all four mutation positive patients. Clinical findings related to HPP, and the genetic test results of four mutation-positive patients have been given in Table 3.
The genomic ALPL variants found in the study group and their pathogenicity scores.
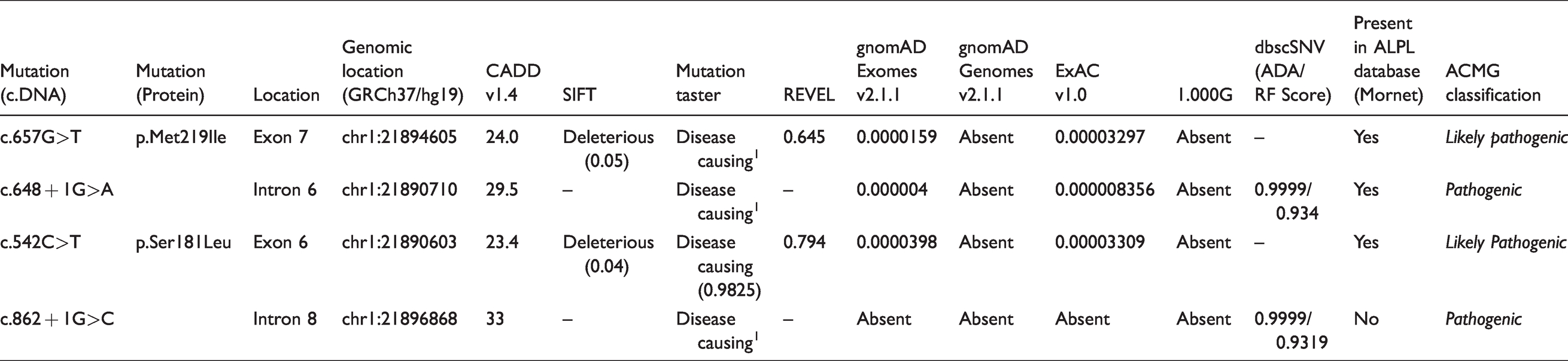
Clinical and molecular findings of four mutation-positive patients.
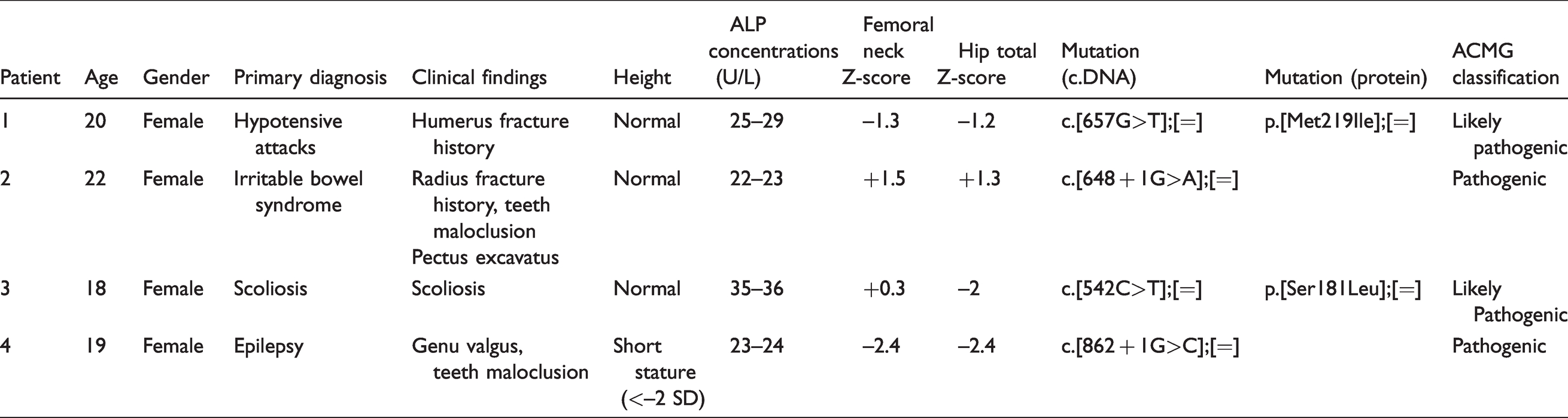
ALPL gene was sequenced in both parents of all patients: The father of Patient 1 carried the mutation. He had a history of nephrolithiasis as an HPP-related symptom. Otherwise, he was normal. Patient 2 inherited the mutation from her mother whose medical history was normal. The father of Patient 3 was carrier and had a history of nephrolithiasis. ALPL sequencing was found to be normal in both parents of Patient 4.
Discussion
The clinical and molecular analysis results of 30 probands having persistently low ALP concentrations (<40 IU/L), were evaluated in this study. ALPL mutations were identified in four individuals (13%). Tenorio et al. performed ALPL mutation analysis in 83 individuals with clinical and biochemical findings suggestive of HPP. Mutations were found in 43.3%. 15 Their cohort, however, included only patients whose original diagnosis was HPP. Moreover, the group also included 17 patients with perinatal/infantile form of HPP and 13 patients with childhood HPP, increasing the expectation of a high mutation positivity rate. 15
In another two studies which included 16 and 50 patients, and were conducted in adult populations with ALP concentrations <30 IU/L in repetitive measurements, ALPL mutations were detected at 43.7% and 87%, respectively.16,17 It has been considered that the lower rate of mutation-positive patients in our study may be due to the selection of patients with higher ALP concentrations. Additionally, our study included only adolescents and young adult patients. Regarding to the results obtained from adult studies, our study may be indicative of low ALP concentrations in young people being influenced more by other factors beyond mutations in the ALPL gene.
In this study, four mutation-positive patients showed the mildest symptoms of HPP. In three of them, the main complaint was not HPP related. Only Patient 3 was admitted to the hospital due to scoliosis. Tenorio et al. reported in adult HPP patients, symptoms usually appeared after 18 years of age. 15 Due to our patients being a young age, they may not yet have developed the clinical findings seen at more advanced age. However, the possibility of them carrying a recessive mutation that is not responsible for HPP symptoms cannot be excluded.
Additionally, in our study, skeletal and dental findings were observed in both mutation-positive and negative patients. As mentioned in the introduction, diagnosis of HPP is challenging especially in mild HPP. In mutation-negative cases, factors that cause low ALP concentrations may be the underlying cause of these clinical findings, or there may be undetected mutations in the HPP gene, such as deep intronic mutations. Therefore, we cannot say these findings are specific for HPP and they also can be incidental. However, as suggested in the previous studies, in cases with low ALP, when a mutation in the HPP gene is detected, these findings should be considered as early signs and followed up.3,15
To date, 400 mutations have been reported in the ALPL gene mutation database. 7 The majority of them are missense mutations with a frequency of 70%. The second most common mutation type is small deletion (11.8%), followed by splicing mutations (5.8%). 7 In the present study, two of four mutations were splicing mutations, the other two being missense mutations.
The mutation c.657G>T (p.Met219Ile), which was detected in Patient 1, is located at exon 7 and had already been recorded in ALPL mutation database. Although, in this database, the clinical picture, caused by it, in a heterozygous situation, has been described as adult HPP, no published article explaining clinical details could be found. Its population frequency has been given as 0.0000159 in gnomAD. 18 No homozygous individuals for this mutation have been reported. Our patient carrying this mutation had a bone fracture as a result of a fall during infancy. Her DEXA showed a low BMD (Table 3). It has been considered that this patient should be followed up, for more prominent HPP findings in older ages.
Patient 2 carried a previously described splicing mutation, c.648 + 1G>A. This mutation was first described in compound heterozygous form associated with a missense mutation p.Gly120Arg in lethal HPP. 19 Later, Sergi et al. reported another lethal HPP case with the same splicing mutation associated with another missense mutation (p.Asn417Ser). 20 In this case, the mutation c.648 + 1G>A was inherited from the father. The carrier father had low ALP concentrations. However, his detailed clinical examination findings had not been reported. An unpublished adult HPP case carrying c.648 + 1G>A and p.Glu191Lys mutations as compound heterozygous, was also mentioned in their report (discussion section). 20 Taking those cases into consideration, Sergei et al. suggested that this mutation causes severe HPP when it is associated with severe mutations, whereas milder clinical findings emerge when it is associated with mild mutations. Our 22-year-old patient carrying this mutation experienced radius fracture after a minor trauma and had mild clinical findings such as pectus excavatum, teeth malocclusion, which supports the idea that, in heterozygous case, it manifests with milder symptoms. It can be hypothesized that the mutation c.648 + 1G>A causes defective splicing of exon 6, resulting in an absence or instability of protein product. Thus, in cases carrying this mutation on both their alleles, severe HPP is expected. However, to date, no such case has been reported.
In our Patient 3, a missense mutation, c.542C>T (p. Ser181leu), was detected on one of the ALPL alleles (Table 3). This mutation, in heterozygous situation, had previously been described in a 15-year-old patient presenting with subnormal serum alkaline phosphatase concentrations, dwarfism and symmetrical bowing arms and legs. 21 However, in that particular study, authors performed co-transfections of wild-type and mutated cDNAs in COS1 cells and showed that this mutation had no dominant negative effect. They suggested that, in the affected cases, there might be a second undetected intronic mutation, or another heterozygous mutation having trans effect on the ALPL mutated allele, in another gene. 21 Spentchian et al. investigated large ALPL gene deletions and reported an infantile HPP caused by this mutation (c.542C>T) associated with exon 12 deletion mutation. 22 Their study showed that there may be large deletions in the ALPL gene that cannot be demonstrated by the sequencing. In Patient 3 presented in this study, regarding scoliosis and low Z scores in BMD measurements, it may be considered that there exists another undetected mutation, such as deep intronic or large deletion mutations, associated with the mutation c.542C>T. Another possibility, considering Baldini’s case and our case, this mutation may also cause, respectively, mild clinical findings in the heterozygous state.
Patient 4 had a novel mutation, c.862 + 1G>C, which affects the splice donor site of exon 8. Splicing mutations are expected to have more severe effects on protein function. In support of this, the ALPL homozygous splicing mutations defined so far are mostly associated with lethal or infantile HPP.7,23,24 However, in heterozygous state or associated with mild mutations, they may result in moderate or mild HPP symptoms.7,25 In a previous study, a mutation (c.862 + 5G>A), very close to the mutation detected in our Patient 4, was reported to cause a severe clinical picture in the homozygous state. 26 Clinical findings observed in our Patient 4 supports the idea that this novel mutation may cause mild or moderate HPP in heterozygous state. However, in the homozygous state, similar to other splicing mutations, a severe HPP picture can be expected.
In conclusion, in this study, the identification of four heterozygous ALPL mutations and characterization of their phenotypic findings in the Turkish population contribute to the knowledge of HPP. Among four mutations, one splicing mutation has not been previously described in HPP. As in the other populations, HPP displays allelic heterogeneity in our population. HPP should be considered in the differential diagnosis of patients with low serum ALP concentrations.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Ege University Scientific Research Projects Coordination (grant number 2017-TIP-027).
Ethical approval
The study was approved by the Ege University Hospital Ethics (Date: 11/04/2017, Number: 17–1.1/5).
Guarantor
EI.
Contributorship
Project design: MBA, GA, BB and FO; data collection and clinical evaluation: MBA, TA, GA, and BB; sequence analysis: BA, EI, and TA.; preparation of the manuscript: MBA, EI, and F.O. All authors reviewed and edited the manuscript and approved the final version of the manuscript.