
Abstract
A significant increase in the activity of serum alkaline phosphatase is commonly reported in patients on long-term antiepileptic treatment or after any uncomplicated fracture. We report a case of a 35-year-old male patient on five different anticonvulsant medications for treatment of the rare autosomal recessive neurodegenerative disorder, Unverricht-Lundborg disease. He presented with bilateral metatarsal fractures: however, his serum alkaline phosphatase activity remained below the lower limit of reference interval. Biochemical laboratory investigations revealed a longstanding low serum alkaline phosphatase and raised plasma pyridoxal-5′-phosphate concentration. Sequencing of genomic DNA revealed that he is heterozygous for a mutation in the ALPL gene, which is consistent with the diagnosis of hypophosphatasia.
Introduction
Hypophosphatasia (HPP) is a rare inherited genetic condition, with heterogeneous phenotype and variable severity, occurring secondary to inactivating mutations of the tissue non-specific isoenzyme of alkaline phosphatase (TNSALP). 1 Six forms of the disease have been described according to the age at presentation and clinical severity. The earlier the onset, the more severe the condition with the mildest form occurring in adulthood, when patients usually suffer from premature teeth loss with little or no other skeletal disease. 2 The biochemical hallmarks of the disease are: reduced serum alkaline phosphatase (ALP) activity, with an increase in plasma pyridoxal-5′-phosphate (PLP). Molecular analysis of TNSALP gene is sometimes necessary to distinguish HPP from other metabolic skeletal diseases. 3 With the availability of an enzyme replacement therapy for treatment of HPP, it is important that laboratories report reliable age- and sex-related ALP reference intervals to aid in the diagnosis of this very rare genetic metabolic disease. 4
We report here the first case, to our knowledge, of Unverricht-Lundborg disease with a recent diagnosis of adult HPP.
Case presentation
The duty biochemist noted a low serum ALP in a 35-year-old male patient who presented with bilateral metatarsal fractures. The man was known to have Unverricht-Lundborg syndrome, an autosomal recessive progressive myoclonic epilepsy secondary to biallelic mutation of the cystatin B (CSTB) gene. He was on five different antiepileptic treatment including: sodium valproate, piracetam, levetiracetam, clonazepam and zonisamide.
He first presented at the age of 10 with generalized tonic-clonic seizures. Electroencephalography showed photosensitivity, and he was diagnosed with idiopathic generalized epilepsy with photosensitivity. He was started on sodium valproate. At the age of 13, he started to develop myoclonic jerks particularly at night. His MRI brain was normal. His condition deteriorated with time: myoclonic jerks became very frequent and disabling with decline in cognitive function, although retaining full capacity, necessitating the use a wheelchair by the age of 23. Sequencing of the CSTB gene, encoding cystatin B, revealed that he was compound heterozygous for a dodecamer expansion mutation and a point mutation, consistent with a diagnosis of progressive myoclonic epilepsy type 1-Unverricht-Lundborg disease. 5
Serum analysis showed low ALP with raised serum phosphate despite normal renal function (Table 1). He had a history of metatarsal fractures and possible right fifth rib fracture. His medical records revealed low serum ALP (Figure 1) and raised serum phosphate on many separate occasions over the past 16 years. Other biochemistry was normal including thyroid, renal and liver functions except for an isolated rise in serum alanine transaminase secondary to fatty liver. He was on colecalciferol 400 units/calcium carbonate 1.5 g chewable tablets once daily with adequate vitamin D status. During his last hospital admission, CT showed fractures involving the second, third and fourth metatarsal bases and middle cuneiform on the left foot and the second and fourth metatarsal bones of the right foot. Despite his uncomplicated fractures and long-term treatment with sodium valproate, 6 both are known to cause an increase in serum ALP, his serum ALP remained low. No other cause of low serum ALP such as metabolic bone disease, coeliac disease or malnutrition was identified in this patient.
Blood results on admission.
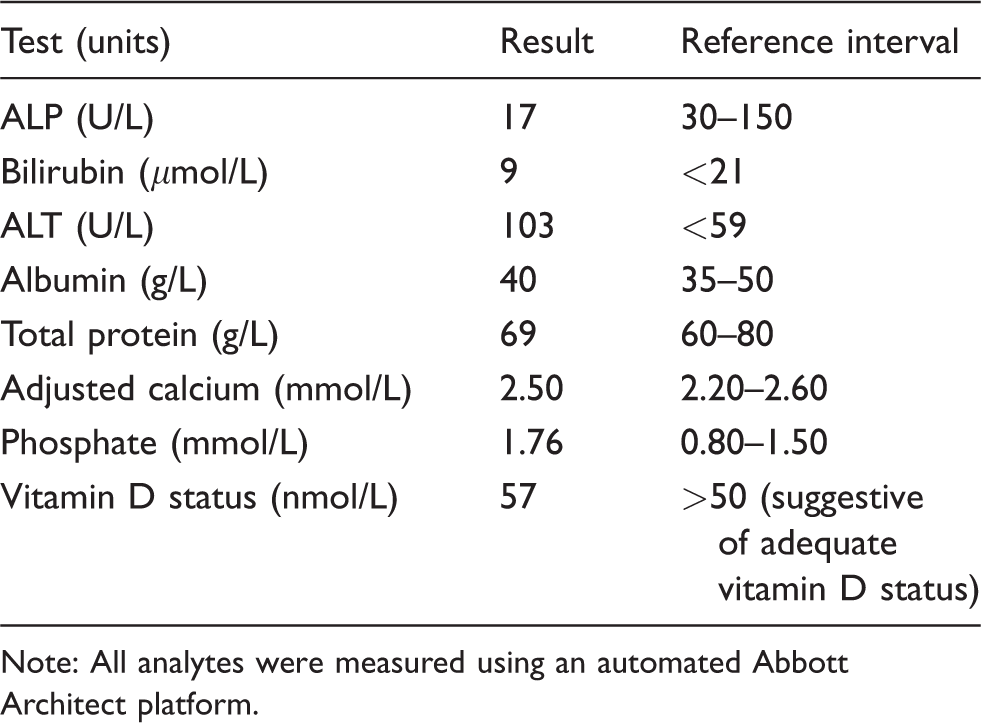
Note: All analytes were measured using an automated Abbott Architect platform.
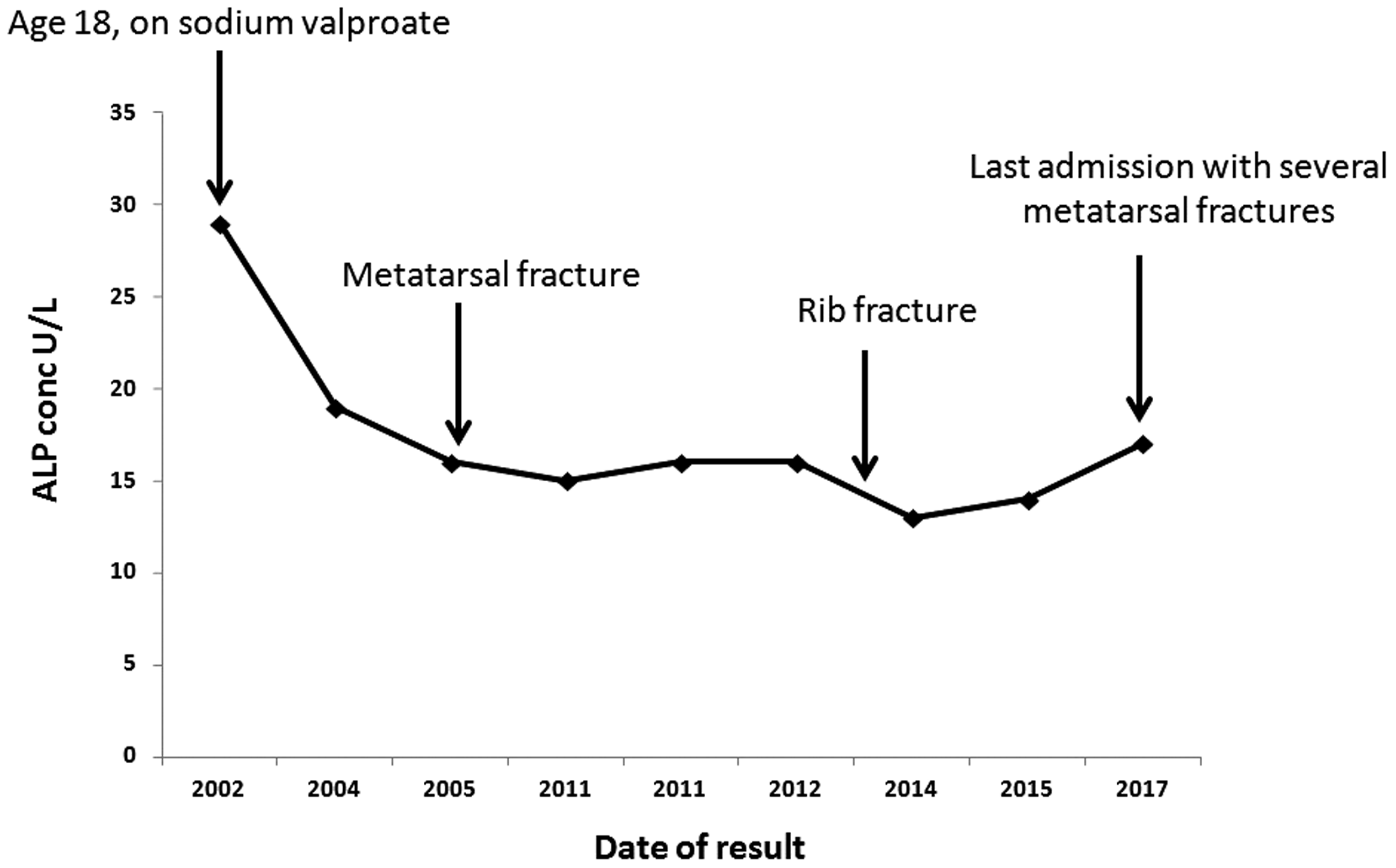
Schematic representation of serum concentration of ALP (U/L) over many years prior to initial presentation.
TNSALP catalyses the hydrolysis of pyrophosphate to inorganic phosphate, the latter crystallizes with calcium, forming the hydroxyapatite required for adequate bone and teeth mineralization. Reduced TNSALP activity results in accumulation of pyrophosphate and an increase in articular calcification, causing joint stiffness and pain. PLP, an activated form of vitamin B6, is another TNSALP substrate which is required as a cofactor in neuronal cells to form neurotransmitters. PLP is dephosphorylated by TNSALP to pyridoxal which then crosses the blood–brain barrier to be re-generated as PLP. Phosphoethanolamine (PEA), though not a confirmed TNSALP substrate, is raised in serum and urine of TNSALP knockout mice, but it is not pathognomic for the condition and its clinical significance is unknown. 7
In view of the history of recurrent fractures and the longstanding low serum ALP, we investigated the possibility of HPP. Plasma PLP concentration was raised (233 nmol/L – reference interval 40–100): he was not taking vitamin B6 supplementation. Urinary PEA was normal. Sequencing of the ALPL gene was performed using the Ion S5 Next Generation sequencing platform (Thermo Fisher Scientific, Loughborough, UK) and the BigDye® terminator purification kit (Thermo Fisher Scientific, Loughborough, UK). The patient was found to be heterozygous for a p.(Ala443Val) c.1328C > T, likely pathogenic mutation 8 in exon 12 of the ALPL gene, which is consistent with a diagnosis of HPP.
Discussion
Serum ALP activity is usually measured to detect an increase in its activity. The clinical significance of the rare finding of low serum ALP activity in the adult population is not universally recognized. Among various causes of low serum ALP is HPP, a rare genetic disorder with a wide spectrum clinical severity and variable expressivity. 9 Six clinical phenotypes have been described based on the age of presentation and the severity of symptoms: perinatal lethal, perinatal benign, infantile, childhood, adult and odontohypophosphatasia. The severe HPP forms (perinatal and most infantile cases) are transmitted in an autosomal recessive manner. The milder forms such as adult and odontohypophosphatasia may be inherited as dominant or recessive traits. 1 ALP is a membrane-bound enzyme that can be classified into three tissue-specific isoenzymes that are intestinal, placental, germ cell and one tissue non-specific ALP (TNSALP) expressed in liver, bone and kidney. Loss-of-function mutations in the gene encoding the TNSALP isoenzyme lead to defective mineralization of the skeleton and teeth. The enzyme is also responsible for dephosphorylation of PLP to pyridoxine, so that it can cross the blood–brain barrier where it is required for the synthesis of gamma-aminobutyric acid (GABA). 7 Some TNSALP mutations are associated with reduced activity to dephosphorylate PLP, leading to reduced GABA synthesis and seizures. 10 Increased urinary PEA concentrations are noted in some patients, but this is not sensitive diagnostic marker for the disease as some patients with HPP have normal PEA excretion.
Adult HPP, typically presenting in middle age, is characterized by foot pain secondary to stress fractures in the metatarsals and is sometimes associated with premature loss of deciduous teeth. Heterozygote carriers usually exhibit some residual TNSALP activity and present with a milder form of the disease. The mutation found in our patient has been previously reported as a dominant negative mutation, and probably accounts for the mild HPP phenotype observed in this case. 11 Our patient had no history of premature loss of deciduous teeth. Bone densitometry was planned but did not take place on this admission as the patient moved out of the area.
Bone remodelling is a dynamic process requiring a balance between bone resorption or osteoclast activity and bone formation or osteoblast activity. Our patient had two genetically inherited conditions that have implications on bone metabolism. He is a compound heterozygote for the rare genetic condition, Unverricht-Lundborg disease. This condition occurs due to mutations in CSTB producing dysfunctional cystatin B, which is a known inhibitor of the lysosomal cathepsins. It has been shown that decreased expression of CSTB mRNA enhances cathepsin activity. 12 CSTB inhibits cathepsin K activity, which is essential for osteoclast function and bone resorption. Patients with Unverricht-Lundborg disease may manifest with bone changes, such as thickening of the skull and long bones, caused by reduced CSTB activity, leading to loss of cathepsin K inhibition and accelerated bone resorption cycle. 13 The exact mechanism, by which lack of CSTB leads to the observed skeletal changes, is not fully elucidated. Interestingly, our patient had no evidence of similar bone changes in any of the skeletal radiographs performed over the years. This might be due to reduced osteoblast-bound TNSALP activity and the associated hypomineralization of bones.
To our knowledge, this is the first case report of a patient who has diagnosis of both Unverricht-Lundborg disease and HPP. Although HPP did not have a significant clinical impact for this patient, further studies would be required to elucidate if PLP might play a role in the management of epilepsy associated with the rare Unverricht-Lundborg syndrome.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Not required. Written informed patient consent has been obtained for publication of this case report.
Guarantor
SZ.
Contributorship
SZ prepared the first draft of the text; all authors reviewed and edited the article and approved the final version for submission.