
Abstract
Background
Detection of disease-associated mutations in patients with familial hypercholesterolaemia is crucial for early interventions to reduce risk of cardiovascular disease. Screening for these mutations represents a methodological challenge since more than 1200 different causal mutations in the low-density lipoprotein receptor has been identified. A number of methodological approaches have been developed for screening by clinical diagnostic laboratories.
Methods
Using primers targeting, the low-density lipoprotein receptor, apolipoprotein B, and proprotein convertase subtilisin/kexin type 9, we developed a novel Ion Torrent-based targeted re-sequencing method. We validated this in a West Midlands-UK small cohort of 58 patients screened in parallel with other mutation-targeting methods, such as multiplex polymerase chain reaction (Elucigene FH20), oligonucleotide arrays (Randox familial hypercholesterolaemia array) or the Illumina next-generation sequencing platform.
Results
In this small cohort, the next-generation sequencing method achieved excellent analytical performance characteristics and showed 100% and 89% concordance with the Randox array and the Elucigene FH20 assay. Investigation of the discrepant results identified two cases of mutation misclassification of the Elucigene FH20 multiplex polymerase chain reaction assay. A number of novel mutations not previously reported were also identified by the next-generation sequencing method.
Conclusions
Ion Torrent-based next-generation sequencing can deliver a suitable alternative for the molecular investigation of familial hypercholesterolaemia patients, especially when comprehensive mutation screening for rare or unknown mutations is required.
Keywords
Introduction
Familial hypercholesterolaemia (FH, OMIM 143890) is the most common monogenic form of hypercholesterolaemia exhibiting an autosomal dominance pattern of inheritance. FH is characterized by elevated serum low-density lipoprotein cholesterol (LDL-C) levels, tendon xanthomas and early onset coronary heart disease.1,2 The prevalence of FH is estimated around 1/500, although recent reports increase prevalence to 1/2003,4 and thus raising a strong possibility of disease under-diagnosis. Early molecular diagnosis of FH in families is extremely important as treatment with cholesterol-lowering drugs can reduce the risk of cardiovascular disease and prolong life expectancy in patients. 5 Genetic epidemiology studies suggest that more than 80% of autosomal dominant FH cases have a causal mutation in the LDLR gene, 6 which leads to defective uptake or processing of low-density lipoprotein particles. 2 Over 1200 different mutations are listed in LDLR gene database of University College London to date. 6 Mutations are distributed throughout the whole gene sequence with no apparent hotspot and include single nucleotide changes, introduction of pre-mRNA splicing defects, deletions and insertions leading to premature stop codons or amino acid substitutions in the LDLR protein and large rearrangements, the latter comprise around 11% of all LDLR mutations. 6 In some ethnic groups, the majority of variants fall within the ligand-binding domain, which is encoded by exons 2 to 6. 7 Disease causing mutations have been also identified in apolipoprotein B (APOB) where the mutational hot spot is at codon 3527 in the LDLR-binding domain of APOB and in proprotein convertase subtilisin/kexin type 9 (PCSK9) genes, these comprise around 5% and 2% of all UK FH mutations, respectively. 8
FH mutation detection poses some considerable methodological and analytical challenges including cost and time efficiency and practicality for screening large number of patients: to address these, different approaches have been developed. These include CE-marked methods that target the most commonly occurring mutations, either using multiplex polymerase chain reaction (PCR) [FH20, Elucigene Diagnostics, UK] or oligonucleotide arrays [LipoChip (Progenika Biopharma) and Randox FH array (Randox Laboratories Limited, Crumlin, UK)] designed to detect 20, 189 and 40 mutations, respectively. The sensitivity of these assays varies and is largely dependent on the number of mutations detected and whether these mutations are represented in the population screened. 9 Conventional capillary (Sanger) sequencing can also provide a more comprehensive sequence coverage with a high degree of accuracy but the requirement for dedicated infrastructure and low throughput capacity limits its practicality. 10 In addition, multiplex ligation-dependent probe amplification analysis (MLPA), a high-throughput and cost-effective method has been shown to be efficient in diagnosing large gene rearrangements. 11
In recent years, next-generation sequencing (NGS) technology has emerged with a capability of massive parallel sequencing of millions of reads. 12 NGS platforms have boosted the throughput and enhanced the sequencing speed and accuracy resulting in reduction in sequencing cost. 10 Moreover, bench-top next-generation DNA sequencers such as Ion Torrent PGM (Life Technologies, NY, US) and MiSeq (Illumina, California, US) have become an attractive option for clinical diagnostic laboratories for applications involving simultaneous targeted re-sequencing of multiple genes. These potential advantages led to the development of various NGS-based methods for detection of LDLR, APOB or PCSK9 mutations in patients investigated for FH.13–17 To test suitability of adoption by frontline non-specialist diagnostic services, we developed and validated a novel NGS-based targeted re-sequencing method in a secondary care NHS biochemistry/molecular laboratory setting. We tested its use in the routine assessment and molecular diagnosis of a small cohort of FH patients exhibiting distinct mutations in LDLR and APOB and compared its performance characteristics to other methodological approaches that offer different solutions to FH patient screening.
Materials and methods
Patient cohorts
The study included DNA samples from 58 patients who attended the University Hospitals Coventry and Warwickshire (UHCW) NHS Trust lipid clinic with a diagnosis of possible FH according to Simon Broome criteria (family history of myocardial infarction and cholesterol levels greater than 7.5 mmol/l at initial presentation) between 2012–2015 and investigated according to routine protocols. Patient characteristics and genomic DNA extraction method are shown in Supplementary Method information. The study was approved by the Arden Tissue Bank Ethics Committee. The flow chart of DNA sample processing according to methods used is shown (Figure 1).
Consort-like flow chart of DNA sample processing according to methods used.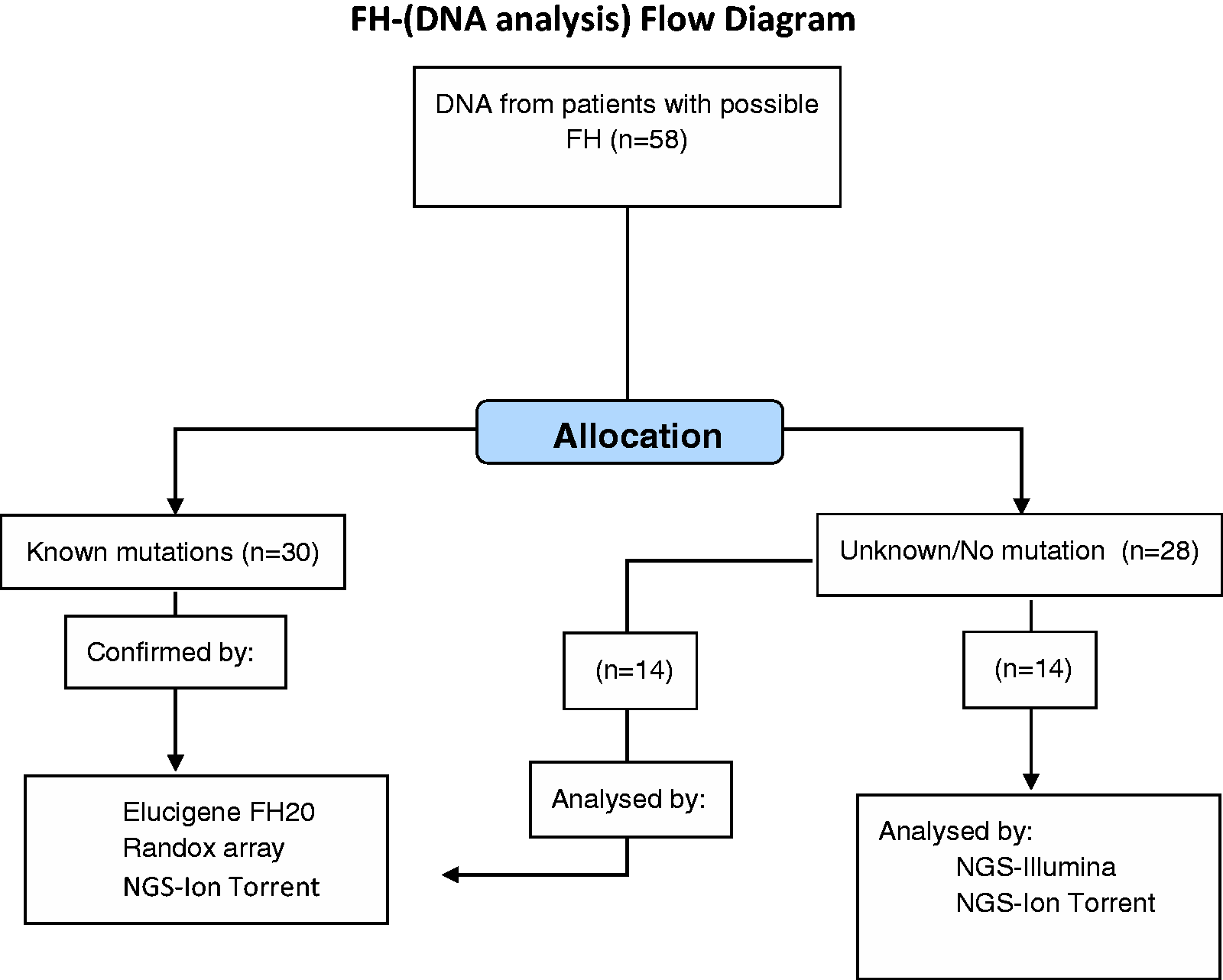
Multiplex PCR assay and FH array
In 44 patients, initial molecular screening was carried out using a targeted mutation amplification refractory mutation system (ARMS)-based multiplex PCR assay (Elucigene FH20 kit). 18 This assay determines 20 most common mutations associated with familial hypercholesterolaemia, 18 mutations in LDLR and one mutation in each of the APOB and PCSK9 genes. Ethidium bromide-stained PCR amplicons separated on 2% agarose gels were visualized under UV transilluminator at 260 nm.
All 44 samples were also subjected to mutational testing using the Randox FH array, a CE-IVD mutation detection assay, which combines multiplex PCR and biochip array hybridization and is designed to detect 40 targets, 38 in LDLR, 10580 G > A in APOB and 1120 G > T in PCSK9 (http://www.randox.com/brochures/PDF%20Brochure/LT367.pdf).
Ion Torrent next-generation sequencing method design and bioinformatics analysis
The NGS method design and characteristics are shown in Supplementary Method information. Primers were designed with the Ion Ampliseq Designer web-based primer design tool and barcoded library was constructed with the Ion Ampliseq Library 200 v2 Kit (Life Technologies, NY, USA). All patient samples were sequenced using 314 v2 barcoded chips. The predicted coverage of known mutations was estimated as 87%. This estimate takes into account that large arrangements (11%) and promoter variants (around 2%) in the LDLR gene would be missed by the NGS method.
The integrated bioinformatics software on the Torrent Server was used for sequencing data analysis. Each run was assessed for pre-alignment and alignment metrics and the sequencing performance was compared between runs. The run was considered successful when>95% of the target sequence was covered at minimum depth of 20×, in agreement with guidelines for NGS analytical performance. 19 The variant caller files (VCFs) were further analysed with the Ion Reporter Software v 4.0. Shortlisted variants were annotated using PolyPhen, sorting intolerant from tolerant (SIFT), Grantham and PhyloP scores and their prior association with FH was assessed using LDLR variation databases.6,20 Mutation nomenclature was based on the Human Genome Variation Society (HGVS) guidelines (http://www.hgvs.org/mutnomen/).
Sanger sequencing
In discrepant samples, LDLR gene exon 4 was amplified with forward (5′ TAGAATGGGCTGGTGTTGGG 3′) and reverse (5′ CCAGGGACAGGTGATAGGAC 3′) primers and exon 10 with forward (5’ACCGTCATCAGCAGAGACAT3′) and reverse (5′CTTCCTGCTCCCTCCATTCC3′) primers. After ExoSAP-IT (Affymetrix Inc., CA) treatment, the samples were sent to GATC Biotech Ltd (The London BioScience Innovation Centre, London, UK) for capillary (Sanger) sequencing. Data were visualized with GATC Viewer Software (GATC Biotech, Germany).
Statistical analysis
Coverage was analysed using the total read count (sum of forwards and backwards read counts) for each amplicon, for each sample. Total read counts were plotted as box plots on a log scale. An axis break has been used to allow the inclusion of total read counts of zero. A dashed line was added to indicate the number of total reads at 20× coverage.
Results
NGS assay performance characteristics
The characteristics of different sequencing runs are provided in Supplementary Table 1. In total, at least 96% bases were aligned to the LDLR reference sequence, 85–89% bases with an AQ20 score (one error per 100 bp) indicating a base call accuracy of 99%. A consistently high coverage of the region of interest was observed across different runs, although there was some variation in coverage between individual samples for all amplicons, which might be related to PCR amplification bias and equimolar pooling accuracy of samples (Supplementary Figure 1). One amplicon targeting 39 bases 3′ of exon 16 of LDLR and 85 bases of intron 16 sequence (genomic position 11238722 to 11238846) had a consistently low coverage (mostly below 20×) across all runs, possibly due to a high GC content (67%) in this DNA region. We observed an excellent uniformity of coverage, which is crucial for successful variant detection. 21 The target base coverage 20× and 100× was at least 96% and 93%, respectively. In order to confirm method reproducibility, three runs SR2, 3 and 6 were repeated yielding the same results.
The NGS assay successfully sequenced all genomic DNA samples from the validation and prospective cohorts that exhibited a wide range of concentrations (8.01–49.66 ng/l) and DNA purity (ratio of absorbance 260/280 1.57–2.21).
Comparison of different mutation-screening molecular approaches in 44 FH patients
Forty-four possible FH patient DNA samples previously screened with the Elucigene FH20 multiplex PCR were analysed with the NGS and Randox FH array methods. A blinded approach was used to analyse all samples and avoid any possible operator bias. The 32 mutation-positive samples included various point mutations (G > A; C > T; T > G; A > G; C > G; G > T; G > C) and one base deletion and most identified mutations were in the coding region of analysed genes except a splice site mutation 313 + 1G > A. The most common mutation identified in this cohort was APOB 10580G > A, with 12 individuals (9 probands) tested positive for this mutation. The most frequently occurring LDLR mutation were 2054C > T and 681C > G (Table 1, Figure 2). The variant caller algorithm detected all previously identified mutations showing 100% and 89% concordance with the Randox array and the Elucigene FH20 assay respectively for genotype. All detected variants occurred in a heterozygous state.
Distribution of FH-associated mutations detected in probands (mutation-positive cohorts). Mutations identified by different methods in the cohort of 44 possible FH patients. Genomic DNA samples extracted from patients. Discrepant results are identified in bold M: male; F: female; C: Caucasian; A: Asian. U: unknown; LDLR: low-density lipoprotein receptor.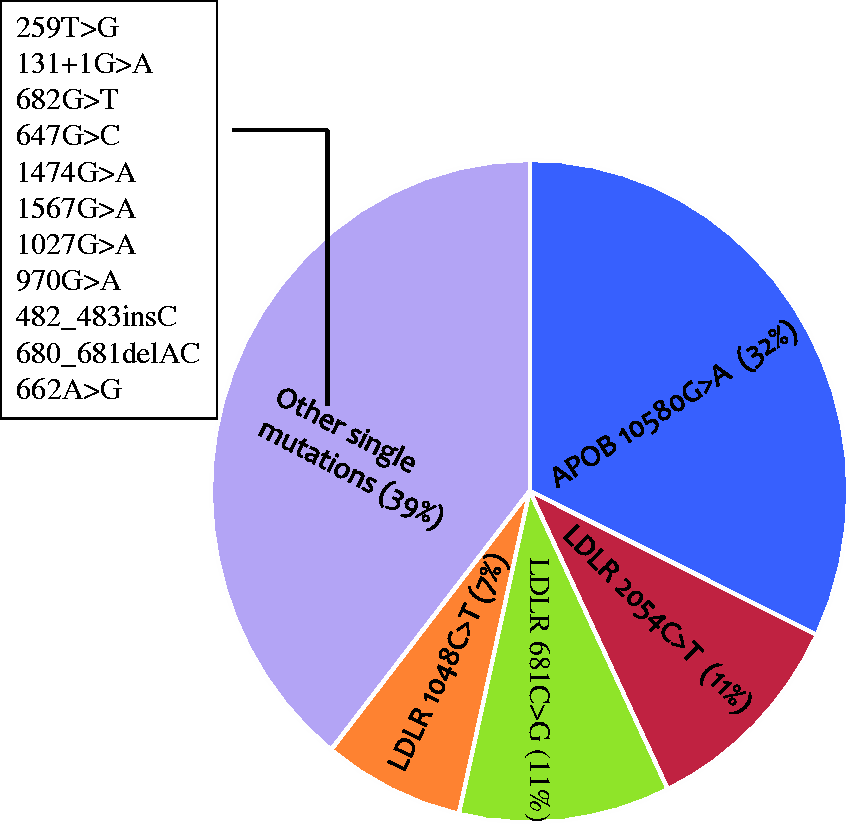

Discrepancies between methods were observed in five samples (two families). The Elucigene FH20 assay detected a LDLR nonsense mutation 682G > T (p.Glu228*) in three members of the same family (FH6, FH7 and FH10), which was not confirmed by the NGS Ion Torrent method or the Randox FH array. Instead a missense mutation c.662A > G (p.Asp221Gly) in the same exon 4, 20 bp upstream of the 682G > T position was identified (Table 1). Figure 3(a) shows the electrophoretic profile of the PCR amplicon from patient FH7 of approximately 164 bp, a size expected for 682G > T LDLR mutation according to the Elucigene FH20 assay manufacturer instructions. Moreover, in another family (sample FH5 and FH11) Elucigene FH20 assay detected presence of a double mutation (681C > G and 680_681delAC). Figure 3(b) shows two DNA fragments of approximately 265 bp (lane 6) and 134 bp (lane 5) indicative of 681C > G and 680_681delAC mutations, respectively, in FH5. Again, this pattern was not confirmed by the NGS assay or Randox FH array; and only the deletion (680_681delAC) in exon 4 was confirmed (Table 1).
(A-B) Agarose gels showing ethidium bromide-stained PCR amplicons obtained by the Elucigene FH20 method that detected (a) the ‘apparent’ c.682G > T (164 bp) mutation of LDLR in DNA sample obtained from patient FH7; and (b) ‘apparent’ c.680_681delAC (134 bp) and c.681C > G (265 bp) mutations of LDLR in DNA sample obtained from FH5. Sanger sequencing profiles of DNA from patient FH7 (c–d) who was identified as heterozygous for the 662A > G mutation and wild type for the 682G > T mutation and patient FH5 (e) who was identified as heterozygous for the c.680_681delAC and wild type for the 681C > G mutation.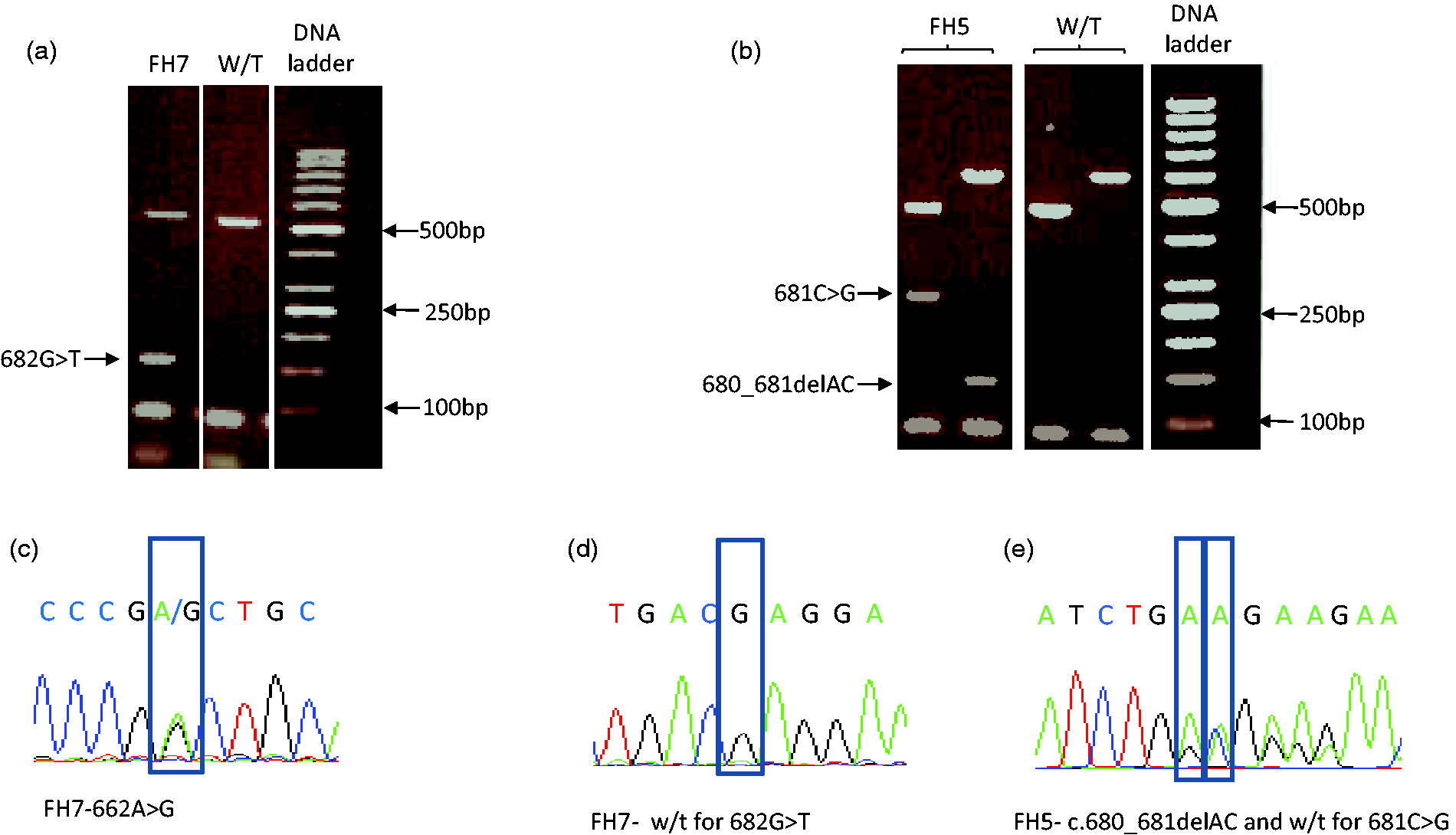
The discrepant results were also evaluated by conventional Sanger sequencing, which also confirmed the NGS and Randox FH array data. Figure 3(c) to (e) shows representative sequencing chromatograms of sample FH7 DNA characterized by presence of 662A > G (p.Asp221Gly) mutation and absence of 682G > T (p.Glu228*) and DNA FH5 positive for the 680_681delAC deletion and negative for 681C > G.
In samples FH31-44, the Elucigene FH20 method failed to detect any mutations (Table 1). These samples were analysed by the NGS Ion Torrent method and the Randox FH array. NGS identified two LDLR alterations (1474G > A and 647G > C) that were not available in the Elucigene FH20 assay and therefore were not detected. The variant 1474G > A (p.Asp492Asn) identified in sample FH31 has been described before in two Taiwanese FH patients but not in normal controls 22 and it is included in the UCL LOVD LDLR database 20 as a probably damaging alteration according to the functional effect prediction tools. This mutation was also detected by the Randox FH array. Furthermore, a non-synonymous variant c.647G > C which leads to a substitution of cysteine to serine at codon 216 was detected in sample FH32 (Table 1). This is a novel alteration that has not been previously associated with FH phenotype. It is not represented in the dbSNP 137 as a common polymorphism (http://www.ncbi.nlm.nih.gov/projects/SNP/). According to PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://sift.jcvi.org/), on-line tools for analysing functional consequences of SNPs, this variant is probably damaging with scores of 1.0 [score ranging 0.0 (benign) to 1.0 (damaging)] 23 and 0.0 (score less than 0.05 are considered deleterious), 24 respectively. The variant c.647G > C is conserved with a PhyloP conservation score of 2.65. Interestingly, a similar modification (G > A) affecting the same nucleotide 647, leading to a Cys to Tyr substitution on codon 216 has been reported in a proband of German origin, where this mutation co-segregated with FH. 25 This cohort did not include any FH cases due to PCSK9 mutations. The DNA analysis of 14 normolipidemic control samples randomly selected did not identify any mutations in the three target genes (data not shown).
Comparison of different NGS methods in 14 FH patients
The Ion Torrent NGS method was used to investigate genomic DNA from 14 possible FH patients screened in parallel for FH-causing mutations by an Illumina NGS platform. The NGS method correctly detected heterozygous alterations in the LDLR in five samples: two missense changes 1027G > A and 970G > A that are included in the UCL LOVD LDLR database 20 as well as three novel variants (Table 1) that included a non-synonymous change c.1567G > C in exon 10 leading to a substitution of valine to leucine at codon 523, a single nucleotide insertion 482_483insC detected in a homopolymer track of 5 cytosines in exon 4 that causes a frameshift and is predicted to create a premature stop codon downstream of the insertion and a synonymous alteration c. 1635G > A (p.Gly544Gly) of unknown significance. The change c.1567G > C is possibly damaging according to PolyPhen-2 (score of 0.657) and SIFT (score 0.01) prediction. The insertion 482_483insC is not included in dbSNP and it is likely to be pathogenic, although family co-segregation studies are required to confirm this. The homopolymer track frameshift mutation 482_483insC was called c.487dupC by Illumina platform, which is likely to be caused by differences in data processing between two NGS platforms (Supplementary Figure 2).
Discussion
Less than 25% of FH cases in the UK and<1% in the USA have their molecular defect identified.26,27 The UK NICE guidelines on FH management strongly recommend identification of causal mutations in suspected cases of FH phenotype followed by cascade testing of first, second and third degree relatives. 28 Genetic testing is considered the most cost-effective detection strategy 29 and for distinguishing monogenic FH from sporadic or polygenic hypercholesterolaemia. 30 However, the spectrum of FH-associated mutations has made screening challenging, especially for frontline clinical diagnostic laboratories that offer molecular testing alongside biochemical assessment of FH patients. As a result, uptake and implementation of these guidelines have been slow due to poor availability, low throughput and high costs of traditional genetic testing, fragmented service delivery and lack of investment in identification of index cases.
We developed an NGS-based solution for comprehensive genetic analysis (CGA)-based investigation and genetic diagnosis of FH and compared performance characteristics with alternative multiplex PCR and array-based approaches. The Elucigene FH20 test was the first commercial assay introduced into routine screening; however, it has a limited sensitivity of between 44% to 52% for patients with a definite FH diagnosis. 31 Moreover, economic evaluation of early efforts to develop targeted tests such as Elucigene FH20 and LIPOChip versus CGA suggest that CGA appears to be the most effective approach in terms of sensitivity and quality-adjusted life years (QALY), 31 although a more up-to-date analysis is required to evaluate the current methods available for screening.
As expected, our study confirmed that Elucigene FH20 fails to identify specific mutations that are not included in the panel of 20 mutations. Although the Elucigene FH20 showed no evidence of false positive results, importantly, we identified two potential issues of mutation misclassification: (a) the c.662A > G (p.Asp221Gly) mutation is misclassified as 682G > T (p.Glu228*) and (b) the presence of 680_681delAC mutation leads to amplification of an additional DNA fragment that is erroneously identified as mutation c.681C > G (p.Asp227Glu). These findings might identify analytical issues of primer specificity and design that might lead to results misinterpretation especially when the test is performed outside a reference laboratory environment. Interestingly, our cohort included patients with ‘true’ 682G > T (p.Glu228*) (n = 1) and 681C > G (p.Asp227Glu) (n = 3) mutations and these were correctly called by all three methods. Nevertheless, these examples of ‘erroneous’ (rather than ‘false’) positivity in FH-associated LDLR mutation detection might introduce confusion and increase possibility of analytical or diagnostic error during the second line cascade testing of relatives.
The Randox FH array showed 100% concordance with the NGS approach over the covered mutations. The FH-associated mutations differ between European countries; 32 UK has one of the most heterogeneous populations with at least 200 different mutations detected 20 and considerable variability in mutation distribution between different UK regions. 33 In our cohort, the NGS method identified novel variants such as c.647G > C and 482_483insC. Similar studies in Saudi patients using the Ion Torrent platform also identified numerous novel variants in the LDLR, APOB or PCSK. 34
The NGS method did not identify any pathogenic mutations in the LDLR, ApoB or PCSK9 in 21 patients with possible FH. Several reports suggest that in some hyperlipidaemic patients the disease is polygenic with a cumulative effect of several LDL-C raising alleles leading to the increased LDL-C.35,36 It is also possible that the genetic abnormality in these individuals is located outside the protein coding region, for example, in the promoter area or deep in the intronic sequence of the LDLR. Such areas were not included in the panel design, as it would require the use of online bioinformatic algorithms and functional studies to determine the effect of the intronic variants on splicing followed by mRNA analysis. A limitation of the current panel design is its inability to detect large rearrangements in the LDLR gene, which might represent an underlying defect in up to 11% of FH cases. 11 To detect this type of sequence alteration, different methodologies such as multiplex ligation-dependent probe amplification (MLPA) are required, although this is also possible by employing recent semiconductor sequencing approaches. 37 Thus, subsequent versions of NGS-based panel could be adapted to detect large insertions and deletions in the LDLR gene to improve accuracy of detection. Nevertheless, our results raise an important point that NGS methods targeting specific genes might not always be able to identify the disease-causing mutation.
Similar to previous reports,37–39 the mean coverage of barcoded samples in the same run was not always uniform, which could reflect variations in sample preparation and quantification or inconsistent performance of NGS across GC-rich DNA regions. 40 This is less likely to be of clinical relevance since the affected region that contains a proportion of exon 16 and intron 16 is characterized by a lack of substitutions (around 2% of all LDLR mutations) 6 and the majority of this sequence is intronic.
In summary, we developed an Ion Torrent-based NGS method for the molecular investigation of patients with FH. Validation of this method in a small cohort of West Midlands-UK possible FH patients identified a number of rare or novel LDLR mutations. Despite the complex technology and limited number of patients tested, this methodological approach supports the NGS potential utility in frontline clinical diagnostic services outside specialist genetics laboratories, when comprehensive sequence coverage of the LDLR gene sequence is required. Our comparison data with other CE-marked assays showed that both NGS and the automated PCR and biochip array hybridization methods were able to analyse successfully DNA samples even with below optimal levels of purity and nucleic acid concentration. These diagnostic assays would allow clinical laboratory services to implement national guidelines according to local population characteristics and offer molecular support in close proximity to the clinic. They also offer methodological flexibility to tailor diagnostic protocols according to methodological complexity, access to specialist skills (i.e. bioinformatics), available resources, ease of use, time efficiency and capacity for high-throughput.
Footnotes
Acknowledgements
The authors would like to thank the UHCW NHS Trust nursing and laboratory staff for their support.
Declaration of conflicting interests
MC, MJL, TLW are employees of Randox the manufacturer of one of the test arrays used in the study. There are no other competing interests.
Funding
This work was supported by a UHCW NHS Trust Research Development and Innovation (RDI) Award and by the Knowledge Transfer Partnerships programme of Innovate UK.
Ethical approval
The study was approved by the Arden Tissue Bank Ethics Committee (12/SC/0526).
Guarantor
Professor Dimitris Grammatopoulos.
Contributorship
All authors made a substantial contribution to the concept and design, acquisition of data or analysis and interpretation of data, drafted the article or revised it critically for important intellectual content, and approved the submitted version.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.