
Abstract
Introduction
Astaxanthin (ASX) is carotenoid with the highest antioxidant activity in various cell types and reverse atherosclerosis. However, the roles and detailed mechanisms of ASX in atherosclerosis associated endothelial injury remains unclear.
Methods
In vitro atherosclerosis model was established in HUVECs by incubation with oxidized low-density lipoprotein (ox-LDL). Cell viability and oxidative stress were measured. The mRNA and protein expressions of lectin-like ox-LDL receptor (LOX-1) and other related genes were determined.
Results
ox-LDL reduced cell viability of HUVECs, and induced oxidative stress, as evidenced by elevated cellular malondialdehyde (MDA) and decreased superoxide dismutase (SOD). Pretreatment with ASX (50, 100, 200, and 400 μM) markedly reversed the reduction in cell viability and an increase in migration of HUVECs induced by ox-LDL (50 μg/mL). ASX attenuated the increase in the endothelial-to-mesenchymal transition (EndMT) process, as evidenced by increased CD31 and decreased α-SMA and vimentin proteins by ASX treatment in HUVECs. Furthermore, ASX attenuated the increase in MDA and decrease in SOD induced by ox-LDL, increased supernatant NO production, attenuated the increase in iNOS and decrease in eNOS in HUVECs with ox-LDL. ASX enhanced mRNA and protein expressions of the lectin-like ox-LDL receptor (LOX-1), which was dependent on ASX’s antioxidant activity. The inhibitory effect of ASX on EndMT could be abolished by overexpression of LOX-1 in HUVECs induced by ox-LDL.
Conclusions
Our data speculate that ASX prevents ox-LDL-induced endothelial cell injury and EndMT by inducing antioxidant property (SOD and NO) and decreasing LOX-1 expression.
Keywords
Introduction
Atherosclerosis is a cardiovascular disease with characteristic progressively accumulated lipid plaques in the aorta. Endothelial dysfunction is a critical pathological process underlying the early initiation and development of atherosclerosis. 1 Endothelial dysfunction can be induced by many risk factors, and oxidized low-density lipoprotein (ox-LDL) is the most important one. 2 Patients with atherosclerosis have dyslipidemia and an increased level of serum ox-LDL, 3 and ox-LDL further induces injury and apoptosis of vascular endothelial cells by facilitating oxidative stress. 4 Oxidative stress also enhances the production of superoxide anion within the vascular wall, thus promoting LDL oxidation, results in a large amount of ox-LDL, and leads to further damage to endothelial function. 5 Therefore, the protection of vessels against oxidative attack is a promising therapeutic strategy for preventing and treating atherosclerosis.
Endothelial cells could transform into myofibroblast-like cells by an endothelial-to-mesenchymal transition (EndMT). 6 During EndMT process, the expression of a series of related genes is altered, with declined endothelial markers (E-cadherin, CD31) and elevated mesenchymal markers (N-cadherin, fibronectin, α-SMA, vimentin). 7 EndMT is also a contributing factor to atherosclerosis. 8 For instance, EndMT could be induced in human aortic endothelial cells through ox-LDL and its receptor, lectin-like oxLDL receptor (LOX-1). 9 However, it remains unclear about the role and related mechanisms of ox-LDL in EndMT.
Astaxanthin (ASX) is a lipophilic carotenoid with inhibitory activities in oxidative injury and inflammation. Astaxanthin can alleviate atherosclerosis risk factors, including antioxidant, antihyperlipidemic, and antiatherosclerotic properties in high fat and high cholesterol diet rats.10,11 One of the mechanisms by astaxanthin is to enhance reverse cholesterol transport, a process of efflux of cholesterol from macrophage-derived foam cells, which is believed to be a primary atheroprotective activity HDL. 12 Currently, astaxanthin has been shown to suppress endothelial injury induced by glucose fluctuation and homocysteine,13,14 but it remains unclear in endothelial cells of atherosclerosis.
In this study, an in vitro model of atherosclerosis was established by incubating endothelial cells with ox-LDL. The effects of astaxanthin on endothelial injury and migration were investigated. We also investigated the underlying mechanism of astaxanthin by studying ROS production, nitric oxide (NO), EndMT, and LOX-1 expression in endothelial cells.
Methods
Cell culture
Human umbilical vein endothelial cells (HUVECs) (Chinese Academy of Sciences, Shanghai, China) were cultured in DMEM (low glucose) with 10% FBS (Invitrogen, Carlsbad, CA, USA), and cultivated in 5% CO2 at 37°C with the humidified atmosphere. HUVECs were incubated with 10, 20, 50, 100, and 250 μg/mL of ox-LDL (Union-Bio Technology, Beijing, China) for 24 h. Before ox-LDL (50 μg/mL) incubation, HUVEC cells were pre-treated with ASX at 0, 50, 100, 200, and 400 μM for 6 h. HUVECS pretreated with 0.1% DMSO served as a control group.
Cell viability assay
HUVECs were cultured in 96-well plates (seeding density: 1 × 103 cells/100 μL), and incubated with various concentrations of ox-LDL and ASX for 24 h. Cells were then washed with PBS and incubated with cell counting kit-8 (CCK-8) solution (1:10 dilution; Beyotime Institute of Biotechnology, Shanghai, China) for 1 h; a microplate reader measured the absorbance 450 nm.
Migration assay
A 24-well Transwell chambers (8 μm diameter) was used to measure the migration capability of HUVECs. Its upper chamber contained 100 μL DMEM medium with 0.1%PBS and seeded with cells at a 1 × 105/mL density. The lower chamber contained a 600 μL DMEM medium with 10% FBS. After 24 h, the more deficient chamber cells were fixed with 95% ethanol for 10 min, stained with 0.1% crystal violet for 30 min, and counted under a light microscope (×100) from five random fields. The migration capability was evaluated by the percentage of migrated cells to total cells.
Measurement of intracellular ROS
HUVEC cells were incubated with DCFH-DA (1:1000) at 37°C in the dark for 1 h. Cells were washed three times with PBS, and a microplate reader measured the absorbance at 485 nm (excitation) and 535 nm (emission).
Determination of MDA, SOD and NADPH oxidase activity
At 24 h of various treatments, HUVEC cells were harvested to extract protein. SOD (Cat no. A001-1; Nanjing Jiancheng, Nanjing, China) was measured at 520 nm absorbance by a microplate reader, MDA (Cat no. A003-1) was measured at 532 nm absorbance, and NADPH oxidase (Cat no. A127) activity at 340 nm absorbance.
Measurement of cellular NO production
At 24 h of various treatments, HUVEC cells were incubated with a NO assay kit (A012-1-2; Nanjing Jiancheng) at 37°C in the dark for 1 h. In this method, nitrate reductase is used to specifically reduce NO3 ¯ to NO2 ¯ and its concentration is determined by colorimetry. Cells were washed three times with PBS, and a microplate reader measured the absorbance at 550 nm.
Exogenous LOX-1 overexpression
Human LOX-1 cDNA was amplified and cloned into a pcDNA3.1 vector. The pcDNA3.1 vector that was transfected with empty DNA served as a negative control. HUVECs were seeded in 6-well plates in complete medium. After growing into sub-confluent (50–60%), cells were infected with pcDNA3.1 vector expressing human LOX-1 (pcDNA-LOX-1, 4 μg plasmid DNA) or empty control (pcDNA) using Lipofectamine 2000 Reagent (Life Technologies, NY, USA). Cells were further cultured in fresh medium for 48 h, and the efficiency of gene overexpression was confirmed by determining mRNA or protein levels.
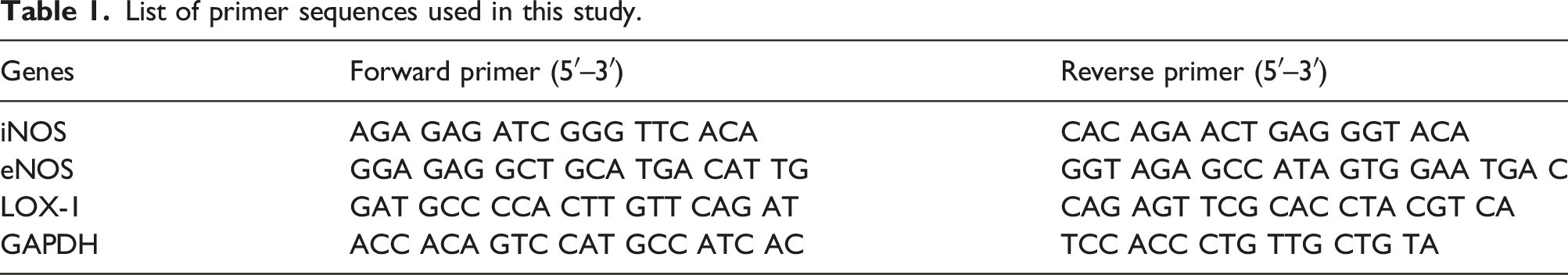
Quantitative real-time PCR
List of primer sequences used in this study.
Western blotting
Total protein was extracted from HUVEC cells. The protein sample (50 μg) was subjected to 10% SDS-PAGE electrophoresis. After transferring to the PVDF membrane, protein samples were blocked with 5% non-fat milk, and was incubated with primary antibodies against CD31 (ab28364, 1:500), α-SMA (ab32575, 1:1000), vimentin (ab92547, 1:1000), LOX-1 (ab214427, 1:500) and β-actin (1:3000; Santa Cruz Biotechnology). After washing, the membrane was incubated with an HRP-conjugated secondary antibody (1:250). Detection of the bands was employed by a chemiluminescent detection system (Thermo Scientific, USA).
Statistical analysis
Data are presented as means ± standard deviation (SD) and analyzed by SPSS 20.0 statistical software. One-way ANOVA was applied to compare the differences between three or more groups, with Bonferroni’s correction for multiple comparison. p < 0.05 was regarded as the criteria for statistical significance.
Results
ASX protected against endothelial injury by ox-LDL in HUVECs
HUVEC cells were incubated with ox-LDL (10, 20, 50, 100, and 200 μg/mL) for 24h to simulate in vitro atherosclerosis model. The cell viability of HUVEC was decreased in a concentration-dependent manner (Figure 1(a)). ox-LDL also enhanced oxidative stress in HUVECs, with markedly decreased SOD and increased MDA (Figure 1(b) and 1(c)). Therefore, 50 μg/mL was used as the optimal concentration of ox-LDL in further experiments. HUVEC cells were pretreated with ASX at 50, 100, 200, and 400 μM for 6h and then incubated with ox-LDL (50 μg/mL) for a further 24 h. ox-LDL treatment for 24 h demonstrated reduced cell viability, and pretreatment with ASX markedly increased cell viability at 100, 200, and 400 μM (Figure 1(d)). ASX suppressed the ox-LDL-induced endothelial cell injury. (a) Cell viability is assessed by CCK-8 assay. SOD activity (b) and MDA level (c) in HUVECs are determined by chromometry. (d) HUVECs were pretreated with ASX (50, 100, 200, and 400 μM) for 6 h and then incubated with ox-LDL (50 μg/mL) for 24 h. ASX attenuates the ox-LDL-induced decrease in cellular viability of HUVECs. Data are expressed as Mean ± SD from at least three independent experiments. *p<0.05, **p<0.01, ***p<0.001 vs. control group; ##p<0.01, versus ox-LDL group.
ASX inhibited migration and EndMT in HUVEC cells with ox-LDL
HUVEC were incubated ox-LDL (50 μg/mL), or ox-LDL plus ASX (50, 100, 200 and 400 μM) for 24 h. A morphological change was observes in HUVEC, from epithelioid to elongated and spindle-shaped fibroblast-like appearance, which can be reversed by ASX (Figure 2(a). Transwell assay was carried out to explore the migration ability of HUVECs. Representative images showed that the migrated cells were stained with blue. Several migrated cells were low in the control group and high in the ox-LDL group and were gradually reduced by ASX treatment (Figure 2(b)). Quantitative analysis showed ASX markedly elevated the percentage of migrated cells in ox-LDL-treated HUVEC cells at all concentrations (p<0.05) (Figure 2(c)). Effect of ASX on the migration and EndMT of HUVECs. (a) The morphology of HUVEC incubated with ox-LDL and ASX. (b) Transwell assay was used to show the migrated cells with 0.1% crystal violet staining, and their representative images are shown (magnification ×100). (c) Quantitative data show that ox-LDL significantly reduced the percentage of migrated HUVEC cells, but ASX significantly increased the percentage of migrated cells. (d) Western blot was applied to measure protein levels of CD31 (e), α-SMA (f), and vimentin (g). ***p<0.001 vs. control; #p<0.05, ###p<0.001 vs. ox-LDL.
ASX suppressed EndMT in HUVECs with ox-LDL
To investigate the effect of ASX on EndMT, western blotting was performed to measure the protein expression of EndMT biomarkers, including CD31, α-SMA, and vimentin (Figure 2(d)). The results showed that ASX significantly decreased protein levels of CD31 (endothelial marker) and increased levels of α-SMA and vimentin (mesenchymal markers) compared with HUVECs with ox-LDL alone (Figure 2(e) to (g)). These results suggest that ASX shows suppressive effects on the process of EndMT in ox-LDL-induced HUVECs.
ASX inhibited oxidative stress in ox-LDL-induced HUVEC cells
To explore whether ASX affects oxidative stress of HUVECs, intracellular ROS, NADPH oxidase activity, and MDA were measured. ox-LDL significantly increased the intracellular ROS (Figure 3(a)), NADPH oxidase activity (Figure 3(b)), and MDA content (Figure 3(c)) of HUVEC cells, which were all significantly decreased by ASX (200 μM) (p<0.05). Therefore, ASX shows a suppressive effect on ox-LDL-induced oxidative stress in HUVEC cells. Modulation of ASX on ROS and NO of ox-LDL-induced HUVECs. (a) ASX attenuates intracellular ROS production. (b) ASX attenuates the activity of NADPH oxidase in HUVECs exposed to ox-LDL. (c) ASX attenuates the increase in MDA of HUVECs exposed to ox-LDL. (d) ASX enhances NO production in HUVECs with or without ox-LDL. The mRNA levels of iNOS (e) and eNOS (f) were measured by real-time PCR. **p<0.01, ***p<0.001 vs. control; ###p<0.001 vs. ox-LDL.
ASX enhanced NO production in HUVECs
We further measured NO content in the culture medium. ox-LDL significantly decreased NO production in HUVEVs (p<0.05). ASX not only enhanced NO production in HUVECs but also reversed the ox-LDL-induced decrease in NO level (Both p<0.05) (Figure 3(d)). We further investigated two nitric oxide synthases, including iNOS and eNOS, which regulate NO synthesis. ASX significantly inhibited iNOS mRNA expression in HUVECs induced by ox-LDL (Figure 3(e)). Moreover, ASX significantly enhanced eNOS mRNA expression in both normal HUVECs and HUVECs influenced by ox-LDL (p<0.05) (Figure 3(f)).
ASX attenuated the increase of LOX-1 in HUVECs with ox-LDL
The expression of LOX-1 in HUVECs was determined by qRT-PCR and Western blot. Ox-LDL increased LOX-1 mRNA and protein expressions compared to the control (p<0.05) (Figures 4(a) and (b)). However, these effects were reversed by ASX (p<0.05). To investigate ROS’s association with LOX-1 in ox-LDL-induced injury, HUVECs cells were simultaneously incubated with ox-LDL and antioxidant N-acetyl-L-cysteine (NAC) (1 m LOX-1 mediates the protective effects of ASX on ox-LDL-induced endothelial injury and EndMT. LOX-1 was detected by qRT-PCT (a) and western blot (b). HUVECs were incubated with ox-LDL and antioxidant NAC (1 m
Discussion
This study reported that ASX pretreatment prevented the ox-LDL-induced reduction in viability, migration, and EndMT. ASX also reduced ROS production and increased supernatant NO. LOX-1 was increased after ox-LDL treatment while it was decreased by ASX pretreatment. The increase of LOX-1 in HUVECs was dependent on ROS. It can be generated in HUVECs by ox-LDL and suppressed by ASX pretreatment, and that this effect depends on LOX-1. Thus, our study shows that ASX is a new therapeutic agent on endothelial dysfunction and uncovers LOX-1 as its underlying target in suppressing EndMT and atherosclerosis.
Atherosclerosis is initiated and promoted by some important mechanisms, such as oxidative stress, 16 endothelial injuries, 2 and the production of inflammatory mediators. 17 The high concentration of ox-LDL results in atherosclerotic plaque formation via multiple pathways, including endothelial apoptosis and ROS generation.18,19 Moreover, ox-LDL incubation also promoted endothelial injury by enhancing oxidative stress and inflammation. 20 This study confirmed that various ox-LDL concentrations significantly decreased the cell viability of HUVECs, with about 20% reduction by 50 μg/mL of ox-LDL. Moreover, our study showed that ASX could protect against endothelial injury and suppress ROS generation. This in vitro study is by previous in vivo study. Administration of ASX increased serum HDL and reduced triglyceride in subjects with mild hyperlipidemia. 21 This indicates that ASX could reduce the risk of atherosclerosis through hypolipidemic effect, confirmed in atherosclerosis mice that an ASX prodrug (CDX-085) reduced aortic arch atherosclerosis. 22 The antioxidative effect of ASX in our study was also following in vivo study that ASX prevented protein oxidation and enhanced activities of superoxide dismutase and thioredoxin reductase in hypercholesterolemic rabbits. 23 ASX could inhibit LDL oxidation and possibly contribute to the prevention of atherosclerosis. 24 Therefore, it can be speculated that ASX might interfere with ox-LDL, thus reduce the amount and efficacy of ox-LDL. Whether this bias exists in our study needs further investigation.
Furthermore, ASX enhanced NO production and eNOS mRNA expression in HUVECs with ox-LDL. Nitric oxide (NO) is a free radical and vasodilative factor produced mainly by eNOS in endothelial cells, and reduction in NO bioavailability contributes to atherosclerosis. ox-LDL decreased NO content and decreased the mRNA and phosphorylated protein of eNOS in HUVECs.25,26 Our results on NO production by ASX are supported by previous study, which showed that ASX increased the mRNA and protein expressions of aorta eNOS and serum NO content in hyperlipidemic rats. 27 The amplified NO production and enhanced eNOS expression in endothelial cells seem to depend on ROS, as in HUVECs with fluctuating glucose, the phosphorylated eNOS protein level was reduced by ASX or the anti-oxidant NAC. 13 Whether this modulation also applies to HUVECs with ox-LDL remains further investigation.
Our results show the inhibitory effect of ASX on migratory capacity and EndMT of HUVESs with ox-LDL. It remains confusing about the exact role of ox-LDL in endothelial cell migration, and the migration of HUVECs was inhibited or enhanced by ox-LDL in Transwell assay.28,29 Our result shows enhanced migratory capacity by ox-LDL, which may lie in calculating the percentage of migrated cells by excluding the suppressive effect of ox-LDL on migrated cell numbers. This speculation was further confirmed by one report that ox-LDL increased MCP-1 and ICAM-1, which are two adhesion-related molecules. 30 and by our result that ox-LDL promoted EndMT in HUVECs, which is an inducer of cell migration. EndMT is an essential contributor to atherosclerosis, 8 and ox-LDL can induce EndMT process in aortic endothelial cells 9 and HUVECs. 31
Furthermore, ASX inhibited migration and EndMT of HUVECs with ox-LDL, and this suggests that ASX can prevent atherosclerosis by targeting endothelial cells. Though it is the first experimental study on EndMT by ASX, our result was supported by previous reports that ASX inhibited EMT in peritoneal mesothelial cells 32 and pancreatic cancer cells. 33 Since EndMT involves the progression and destabilization of atherosclerotic plaques, fibroblast infiltration, and secretion of matrix metalloproteinase, 34 the detailed mechanisms of ASX on endothelial cells deserve further investigation.
This study shows that ASX attenuated LOX-1 expression in HUVECs with ox-LDL. LOX-1 is an ox-LDL receptor in endothelial cells, and LOX-1 mediates most signal pathways of ox-LDL. LOX-1 plays a vital role in the inflammatory response and lipid deposition of the endothelium.35,36 Our results are by one previous report that ASX decreases LOX-1 expression in the aorta and improved endothelial dysfunction in diabetic rats. 37 Our study indicates that inhibition of LOX-1 is one of the mechanisms by which ASX improves endothelial function.
Moreover, LOX-1 seems to lie downstream of ROS in HUVECs, as LOX-1 protein expression can be reduced by antioxidant NAC and be increased by pro-oxidant tBHP in HUVECs. There is a positive feedback regulatory loop between LOX-1 and ROS. ROS oxidizes native LDL and enhances LOX-1 expression, and LOX-1 activation per se stimulates ROS generation. 38 Therefore, breaking the vicious circle by an antioxidant, such as ASX, could markedly retard the process of endothelial damage by targeting LOX-1. Our results showed that LOX-1 overexpression enhanced vimentin expression in HUVECs with ox-LDL and ASX. This indicates that LOX-1 involves the EndMT process by ox-LDL and the inhibition of EndMT by ASX pretreatment. The protein expression levels of EndMT biomarkers were entirely abolished by pretreatment with LOX-1 blocking antibody in human aortic endothelial cells. 9 The potential mechanism may lie in the induced expression of TGF-β. The binding between ox-LDL and LOX-1 promoted TGF-β expression arterial endothelial cells in vivo 39 and increased collagen production in myofibroblasts, 40 thus eventually facilitate EndMT. AST could inhibit TGF-β1 expression and attenuate liver fibrosis or renal interstitial fibrosis.41,42 The detailed mechanisms underlying EndMT by LOX-1 remains further study.
This study has several limitations. Firstly, only in vitro model of HUVECs was used by ox-LDL induction. Up to now, ASX has shown cytoprotective effects in HUVECs by glucose fluctuations, 13 H2O2, 43 or glycated protein/iron chelate. 44 It remains unclear whether ASX protects endothelial injuries from other risk factors of vascular diseases. Secondly, only oxidative stress and EndMT were investigated in this study, and other mechanisms regulated by ASX should be further explored. Thirdly, the effects of ASX are only confirmed in vitro model. The endothelial protection of ASX and related mechanism should be further validated and studied in vivo models of atherosclerosis.
Conclusions
ASX demonstrates protective activity in ox-LDL-induced HUVECs. The mechanisms may be associated with inhibition of oxidative stress, activation of the eNOS pathway, and suppression of LOX-1 expression. The antioxidant effect of ASX might promote the interruption of the vicious circle between LOX-1, ROS and inhibition of EndMT, thus protecting endothelial cells. This study provides ASX as a promising agent in preventing atherosclerosis.
Footnotes
Author Contribution
ZZ wrote the manuscript; JL and RT performed experiments; XZ analyzed the data and revised the manuscript; BY designed and supervised the study. All authors have read and approved the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was financially supported by The Outstanding Clinical Discipline Project of Shanghai Pudong (Grant No: PWYgy-2018-08).
Ethics approval and consent to participate
This study is based on the cell line. We have obtained a statement from the ethics committee of Shanghai Pudong Hospital that ruled that no formal ethics approval was required in this particular in vitro study.