
Abstract
This review focuses on statistical quality control in the context of a quality management system. It describes the use of a ‘Sigma-metric’ for validating the performance of a new examination procedure, developing a total quality control strategy, selecting a statistical quality control procedure and monitoring ongoing quality on the sigma scale. Acceptable method performance is a prerequisite to the design and implementation of statistical quality control procedures. Statistical quality control can only monitor performance, and when properly designed, alert analysts to the presence of additional errors that occur because of unstable performance. A new statistical quality control planning tool, called ‘Westgard Sigma Rules,’ provides a simple and quick way for selecting control rules and the number of control measurements needed to detect medically important errors. The concept of a quality control plan is described, along with alternative adaptations of a total quality control plan and a risk-based individualized quality control plan. Finally, the ongoing monitoring of analytic performance and test quality are discussed, including determination of measurement uncertainty from statistical quality control data collected under intermediate precision conditions and bias determined from proficiency testing/external quality assessment surveys. A new graphical tool, called the Sigma Quality Assessment Chart, is recommended for demonstrating the quality of current examination procedures on the sigma scale.
Introduction
Since publication in 2003 of a review ‘Internal quality control: planning and implementation strategies,’ 1 quality control (QC) has evolved as part of a comprehensive quality management system (QMS). The language of quality today is defined by International Standard Organization (ISO) in an effort to standardize terminology and quality management practices for world-wide applications. Global guidance is provided by the ISO 15189 document for quality management in medical laboratories 2 and is followed by many countries, though with some exceptions such as the US where the clinical laboratory improvement amendments (CLIA) still govern laboratory quality practices. 3 Even in the US, ISO inspection and accreditation for medical laboratories is provided by organizations such as College of American Pathologists (CAP) and American Association for Laboratory Accreditation (A2LA) , thereby acknowledging the primacy of ISO 15189 guidance for a broad comprehensive QMS.
ISO 15189 provides ‘high-level guidance’, meaning that it identifies concepts and principles for good laboratory practice without prescribing the details for implementation. For example, section 5.6.2.1 on quality control states that ‘the laboratory shall design quality control procedures that verify the attainment of the intended quality of results.’ The emphasis is on design of QC procedures, in contrast to the CLIA prescriptive language that requires laboratories to analyse two levels of controls each day [§493.1256 (d3i)], which is the minimum or default statistical QC (SQC) practice adopted in many US laboratories. The advantage of the ISO language is to point out what should be accomplished (design of QC to verify the intended quality of results) without being limited by current practices (e.g. analyse two levels per day).
‘Verification of the intended quality of results’ should be the driving force for today's quality control practices. This of course requires that the laboratory defines the quality needed for the medical applications of an examination procedure as the starting point for any objective and quantitative quality control procedure, process, or system. The importance of quality specifications has been discussed in a recent review by Plebani:
4
A better analytical quality should be achieved by setting and
These recommendations are further supported by studies that assess the current quality of examinations vs. the quality needed for intended use, such as described by Jassam et al.:
5
Evidence from our data shows that The implementation of national and international guidelines is beginning to standardize clinical practice. However, since many guidelines have decision limits based on laboratory tests, there is an urgent need to ensure that different laboratories obtain the same analytical results on any samples. A
To meet these needs requires both improved examination procedures and improved QMSs. A scientifically based quality control process can be developed based on QMS concepts and Six Sigma principles and tools. A Six Sigma quality management system (6σQMS) is presented here that outlines critical practices for achieving the analytical quality required for intended clinical use.
A 6σQMS
Deming's Plan-Do-Check-Act model (PDCA cycle) provides the fundamental building block for developing, implementing and operating a scientific QMS. Deming assigned management the responsibility for maintaining the balance between the many parts of a production operation and urged that they apply the scientific method (PDCA cycle) to make objective, data driven decisions. 6 Burnett, who chaired the ISO 15189 committee, also adopted the PDCA model in his book 7 on implementing ISO 15189 to promote a process flow understanding of the ISO requirements, rather than a static list of requirements. Likewise, we adopt the PDCA model for development, implementation and operation of a QMS in our own book, 8 where we consider both management and technical requirements.
In the discussion here, we focus on the technical requirements and the integration of Six Sigma concepts and principles to provide a quantitative methodology for managing analytical quality. Figure 1 provides an overview of the 6σQMS. Our purpose here is to emphasize that this QMS system fits the Deming PDCA model and again point out that this is the fundamental building block for any QMS and should be employed in the planning, implementation, and operation of a QMS. There are PDCA cycles within the phases for organizing, developing and implementing management requirements, as well as technical requirements. There are PDCA cycles in operating QMS systems and even for organizing for inspection and accreditation. This PDCA model also applies to the work of project teams and even the work of individual laboratory scientists. Thus, the PDCA model is fundamental to scientific management in all phases and activities in the laboratory.
Plan-Do-Check-Act model for implementing a Six Sigma quality system.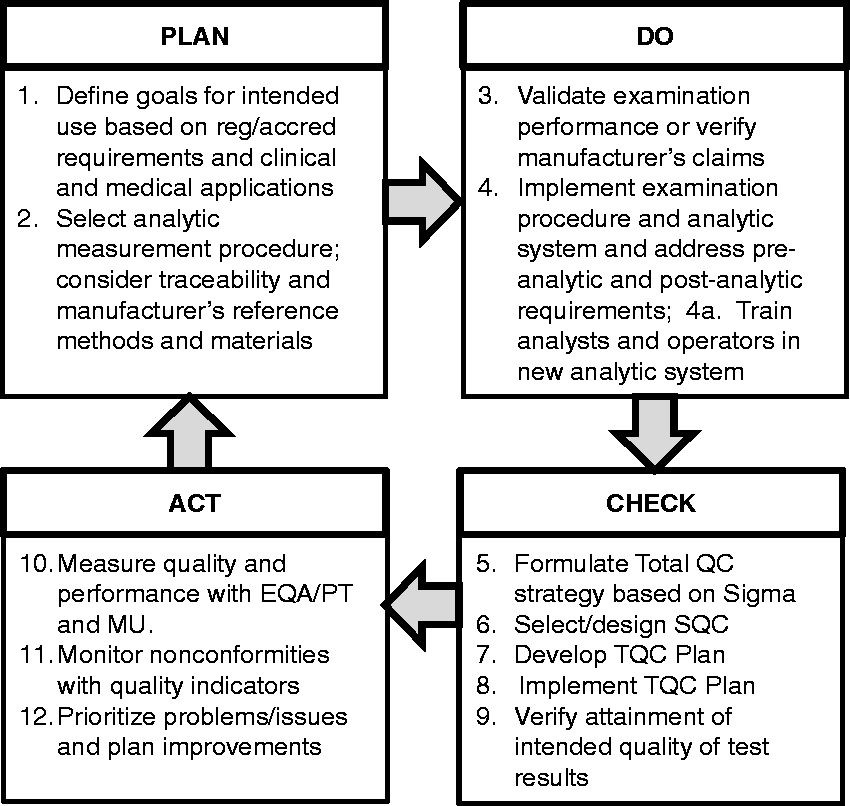
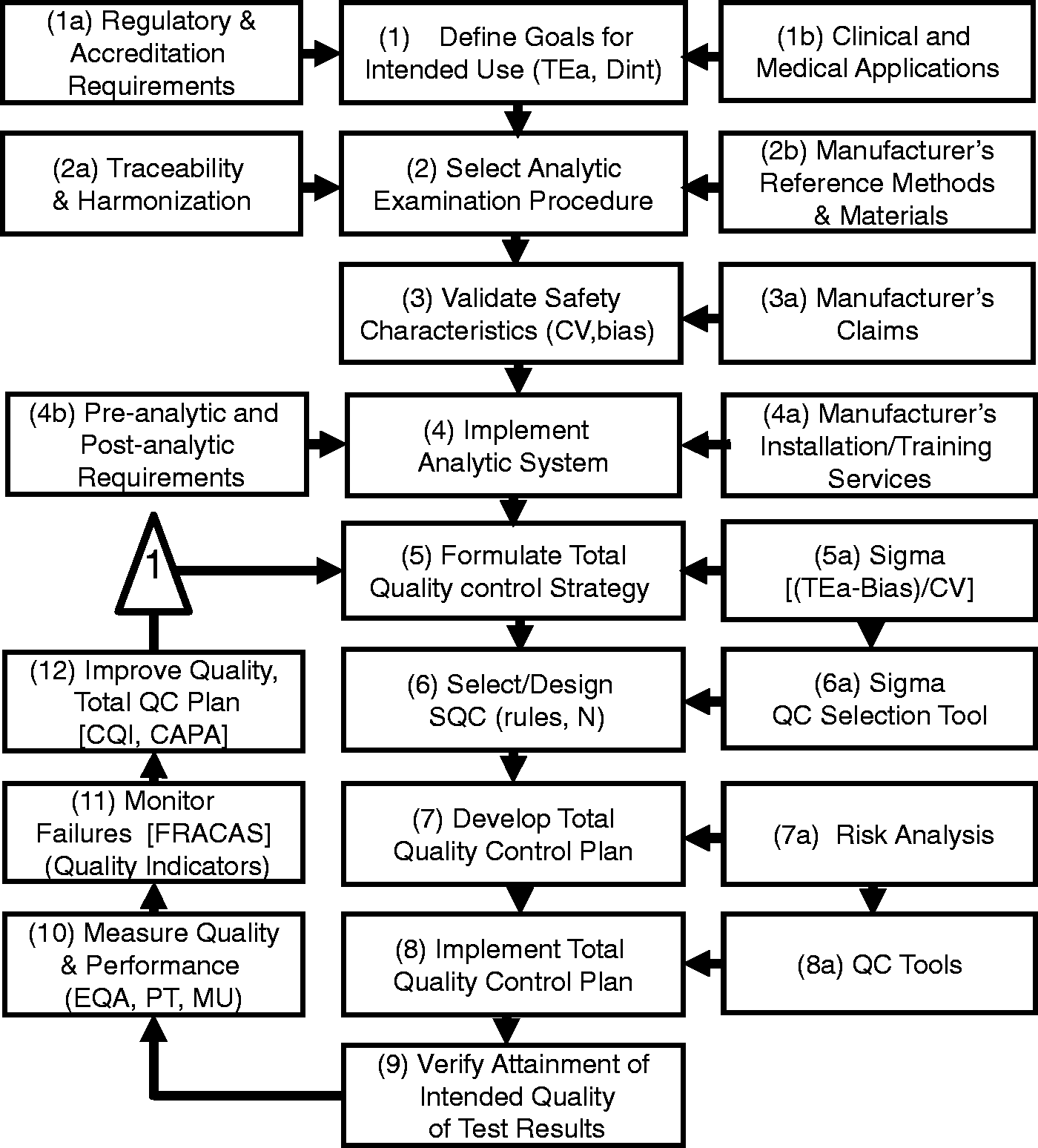
Figure 2 describes a more detailed flowchart of the steps for the 6σQMS and the discussion here is organized around these steps.
The PLAN stage includes steps 1–2 where intended use is defined in terms of an analytical (TEa, allowable analytic total error) or clinical (Dint, decision interval) quality requirement and an examination procedure is selected to satisfy the intended use, with particular attention to traceability or harmonization. The DO stage includes steps 3–4 where the performance of the examination procedure is validated for the defined intended use and a sigma-metric is determined to characterize quality on the sigma-scale. A standard operating procedure (SOP) is developed, analysts are trained and the examination procedure is implemented for routine service. The CHECK stage includes steps 5–9, where a total QC strategy is developed on the basis of the observed sigma quality, an SQC procedure is designed/selected on the basis of the observed sigma quality, and a QC plan is developed to monitor the total testing process, including preanalytic, analytic, and postanalytic phases. The ACT stage includes steps 10–12, where performance and quality are monitored via proficiency testing (PT) or external quality assessment (EQA) programmes, measurement uncertainty (MU) is determined, patient safety is monitored by quality indicators, and improvements are made as needed to the examination procedure and QC plan.
Detailed flowchart of PDCA model for implementing a Six Sigma quality system.
Step 1. Define goals for intended use
Definition of the quality required for the intended clinical use should be the starting point for managing quality! Laboratories still struggle to do this, in spite of more than 50 years of discussion and recommendations in the scientific literature. Confusion persists about the type of requirement that should be defined, e.g. an allowable analytic SD, allowable analytic bias, or allowable TEa. In addition, clinical guidelines may define patient classifications on the basis of medical cut-offs and intervals between different cut-off concentrations, which may provide specifications for a clinical Dint that expands TEa by including other components of variation, such as within-subject biologic variation. 9
HbA1c provides an example of an important measurand for which there are global clinical diagnostic and treatment guidelines, as well as analytical performance specifications. For example, the American Diabetes Association (ADA)
10
recommends classifying patients as follows:
≤5.6 %Hb (37.7 mmol/mol) is considered normal; 5.7 to 6.4 %Hb (38.8 to 46.4 mmol/mol) represents prediabetes; 6.5 %Hb (47.5 mmol/mol) is the cut-off for diagnosis of diabetes; 7.0 %Hb (53.0 mmol/mol) is the target for treatment.
In certifying the ‘equivalence’ of HbA1c measurement procedures, the US National Glycohemoglobin Program (NGSP) 11 specifies a TEa for agreement between a manufacturer's method and the NGSP secondary reference procedures, which is currently specified to be 6.0%. However, earlier the specification was considerably larger (approximately 11%). Likewise, the College of American Pathologists (CAP) criteria for acceptable performance in PT were initially much larger, beginning with 15% more than five years ago, then narrowing to 10%, 7% and currently 6%.
Figure 3 shows a comparison of clinical and analytical requirements using an error grid
12
to demonstrate that by 2014, the certification and PT criteria had been properly aligned to demand the same performance for both the clinical diagnostic and treatment criteria. Nearly two decades of international efforts drove this standardization, alignment of analytical and clinical requirements for quality and improvement in performance of HbA1c. That attests to the difficulties in understanding and defining requirements for intended use.
2014 comparison of HbA1c quality requirements for intended clinical diagnosis and treatment, NGSP certification requirements (Tea = 6.0%), and CAP proficiency testing criterion for acceptability (Tea = 6.0%). Fpos refers to false positive classification and Fneg to false negative classification according to clinical guidelines.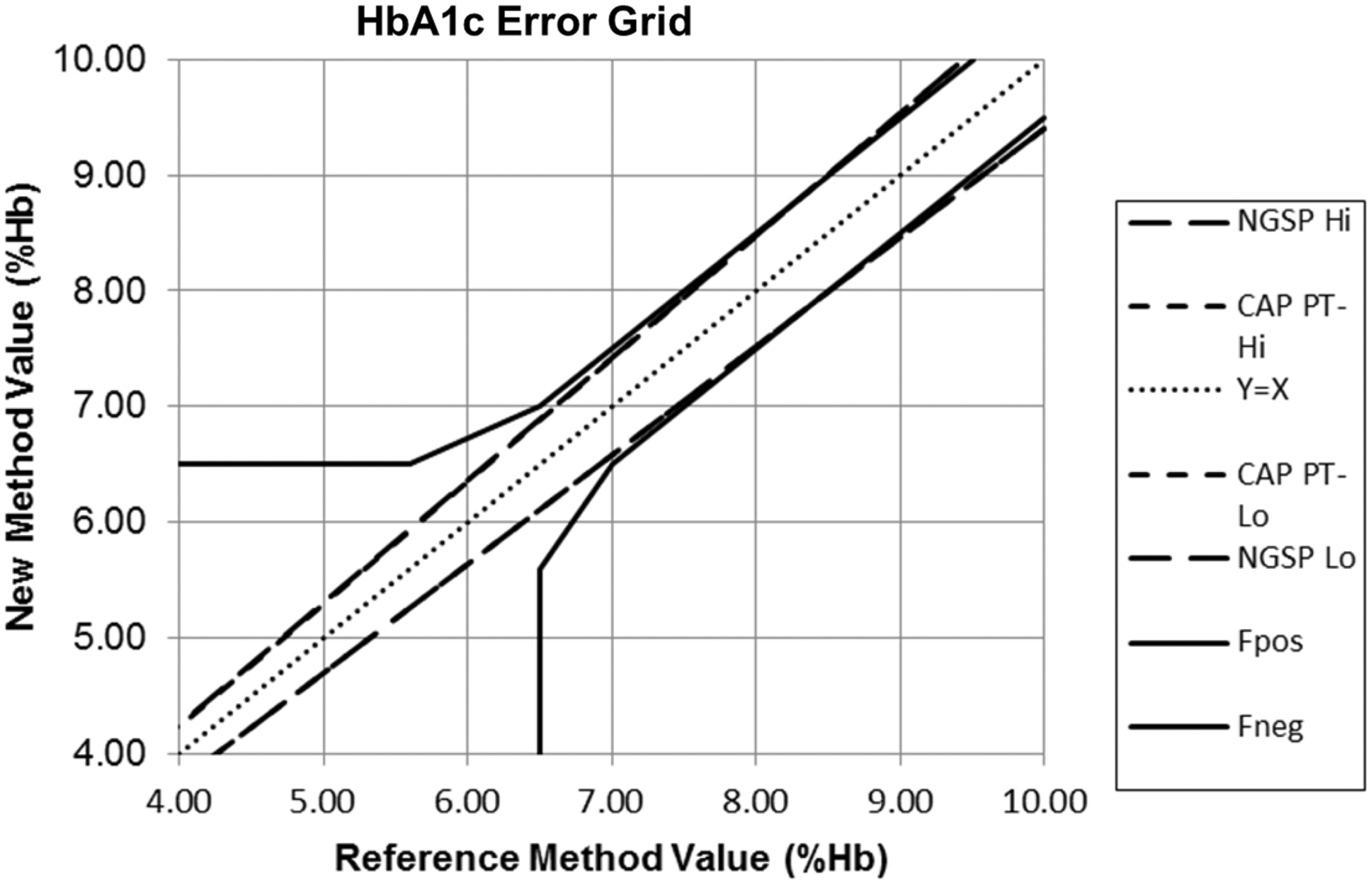
At the time of our earlier review, consensus recommendations for defining quality requirements were available from the 1999 Stockholm conference on ‘strategies to set global analytical quality specifications in laboratory medicine’. 13 More recently, the European Federation for Laboratory Medicine (EFLM) organized a meeting in Milan to review and update the ‘Stockholm guidelines.’ 14 An ongoing task force and several work groups were established to resolve current issues in setting and applying quality goals. The proceedings are available in the May 2015 issue of Clinical Chemistry and Laboratory Medicine. 15
The main revisions simplify the hierarchy from five to three levels, 14 add criteria for review of studies on biologic variation to qualify for inclusion in the database16,17 and provide greater emphasis on separate specifications for imprecision and bias rather than a combined TEa goal. Oosterhuis 18 has criticized the model used for calculating TEa from the equations utilizing biologic variation (CVi, within individual variation; CVg, between individual variation; CVb, overall or total biologic variation) that were first recommended by Fraser and Hyltoft Petersen. 19 An earlier Gowans et al. model 20 that was used to derive the maximum allowable bias (0.25 × CVb) for diagnostic classification assumed a condition where the analytic SD was zero, whereas the Fraser–Petersen model adds the maximum allowable bias for diagnostic classification with the maximum allowable CV for monitoring individual patients (0.5 × CVi) based on within-subject biologic variation. The specific criticism is that there is no theoretical basis for combining a maximum allowable bias with a maximum allowable CV to calculate a biologic TEa goal. All goal-setting models make assumptions and, in this case, the Fraser–Petersen model assumes the most demanding condition for precision (maximum allowable CV for monitoring individual patients) and the most demanding condition for bias (maximum allowable bias for the purpose of diagnostic classifications). That certainly provides a rational basis for the individual specifications for allowable CV and allowable bias. The rationale for further combining those individual specifications to establish a specification for TEa is the laboratory practice of making only a single measurement on each patient sample, which means the patient test result is subject to both random and systematic errors.
In any case, setting a goal for TEa is a different problem from utilizing a TEa criterion for analytical quality management, e.g. to evaluate the quality of a method, to characterize quality on the Sigma-scale, to select SQC procedures and to monitor PT/EQA results. It is important to recognize that the biologic goals found in the Ricos database 21 are currently widely used and frequently more demanding than corresponding goals in PT and EQA programmes. Indeed, a common criticism about biologic TEa goals is the tightness of the goals – for some analytes, TEa is so small there are no methods that can achieve the goal. To overcome that limitation, a new goal-setting model has been proposed by Oosterhuis and Sandberg 22 that mixes biologic goals with state-of-the-art performance. However, that model becomes a lower level 3 model and is inherently flawed because it includes the observed method CV when setting a goal for the allowable CV.
It is critical that the goal-setting process strikes a balance between the most scientifically derived calculations and the practical goals needed for routine operation. It is of no practical help to create a laborious process that generates a metrologically pure quality goal that is so complicated no one can understand its derivation or intended use. It remains to be seen whether the EFLM task forces can balance the metrological complications with the practical simplicity that is needed for adoption and application in medical laboratories. There is a danger that the outcome may revert to the half-century old practice of splitting the quality specifications into separate goals for allowable precision and allowable bias. Again, while it may be easier to scientifically construct separate specifications for precision and bias, the effort ignores the reality that PT and external quality assessment programmes that are required as part of any QMS uniformly employ specifications for TEa that encompass both imprecision and bias. Thus, like it or not, specifications for TEa are needed for the practical implementation of a QMS.
In late 2014 and early 2015, we surveyed 480 medical laboratories (with responses from 80 different countries) to assess current practices in applying different types of quality goals. 23 Some 63% of laboratories indicated their use of goals for TEa, 58% for biologic goals for allowable SD, 47% for use of manufacturers-claimed performance, 38% for use of biologic goals for TEa, 34% for ‘state-of-the-art’ performance, 34% for use of biologic goals for allowable bias, and 32% for goals for clinical intended use. This finding of the high prevalence of TE goals correlates with the fact that most laboratories utilize the goals that are specified in PT and EQA programmes, which they are required to participate for inspection and accreditation. Thus, the practical need in most laboratories is for total error goals, rather than separate goals for precision and bias.
Step 2. Select analytic examination procedure
Selection of analytic systems today is frequently driven by cost considerations, rather than by quality requirements. With the consolidation of testing into multitest analytic systems, there often is a trade-off between cost and quality. Selection of examination procedures is critical to achieve comparability of results within a laboratory, within a healthcare network or system, within a geographic or national region and also globally. Consideration of traceability and harmonization should be a high priority because once the analytic system has been implemented, it is difficult to address issues of biases between examination procedures. According to ISO 15189, medical laboratories should document the metrological traceability of calibration standards on the basis of information provided by the manufacturer, or if necessary, by use of certified reference materials or by comparison with higher order reference procedures.
Traceability is defined as ‘the property of the result of a measurement or the value of a standard whereby it can be related to stated references, usually national or international, through an unbroken chain of comparisons all having stated uncertainties.’ 24 The traceability chain, from the top down, begins with the definition of SI-unit, primary reference measurement procedure, primary calibrator, secondary reference measurement procedure, secondary calibrator, manufacturer's selected measurement procedure, manufacturer's working calibrator, manufacturer's standing measurement procedure, manufacturer's product calibrator, end user's routine measurement procedure, which provides the analysis of a routine sample and produces a test result.
The International Committee of Weights and Measures (CIPM) and the International Bureau of Weights and Measures (BIPM) have created the Joint Committee for Traceability in Laboratory Medicine (JCTLM) to identify, review, and publish lists of higher order certified reference materials and reference measurement procedures. JCTLM maintains databases for reference materials and reference methods that can be accessed at their website www.bipm.org/jctlm.
However, the classical model for traceability is difficult to apply in the measurement of biologic materials. According to Thienpont et al., 25 this model may not apply for many tests in laboratory medicine due to ‘physico-chemical complexity,’ where biologic matrices are complex and influence the analytical results, measurands may be a class of substances rather than a single species, and different measurement procedures sometimes demonstrate specificity to one or a few of the substances in that class. Therefore, other traceability models are needed that begin with international conventional reference measurement procedures, international conventional calibrators, international protocols for value assignment and even manufacturer's selected measurement procedures.
These difficulties can be illustrated for HbA1c, which is well established globally as a key diagnostic and prognostic indicator for patients with diabetes. Figure 4 describes a traceability model in the US, where all HbA1c examination procedures must be certified by the National GlycoHemoglobin Standardization Program (NGSP), which provides a testing service whereby manufacturers can compare their results on a group of 40 patient samples to the results obtained by the US secondary reference methods. Based on such comparison, manufacturers establish a ‘calibration’ for their method to provide test results that are equivalent to the US reference method. Globally, there are two other national standard methods (Sweden and Japan) and an international standard method by the International Federation for Clinical Chemistry (IFCC). All these methods are monitored to maintain consistency through a global network of reference laboratories and calibration materials are developed and tested to maintain the comparability of test results between these established reference methods. International reference materials, along with international and national reference methods, support national calibration services to provide supposedly equivalent test results from routine methods supplied by different manufacturers. In addition, CAP provides special ‘whole blood’ samples for PT to minimize matrix effects and monitor the comparability of methods. Thus, here is a complete system to establish the comparability of test results from different methods together with a PT programme to monitor the achievement of comparability of results.
Traceability chain for ‘physio-chemically complex’ examination for HbA1c. Test results are traceable to an IFCC-reference method via national standardization programmes, such as the US NGSP certification programme. Comparability is monitored through EQA or PT surveys, e.g. the College of American Pathologists (CAP) programme.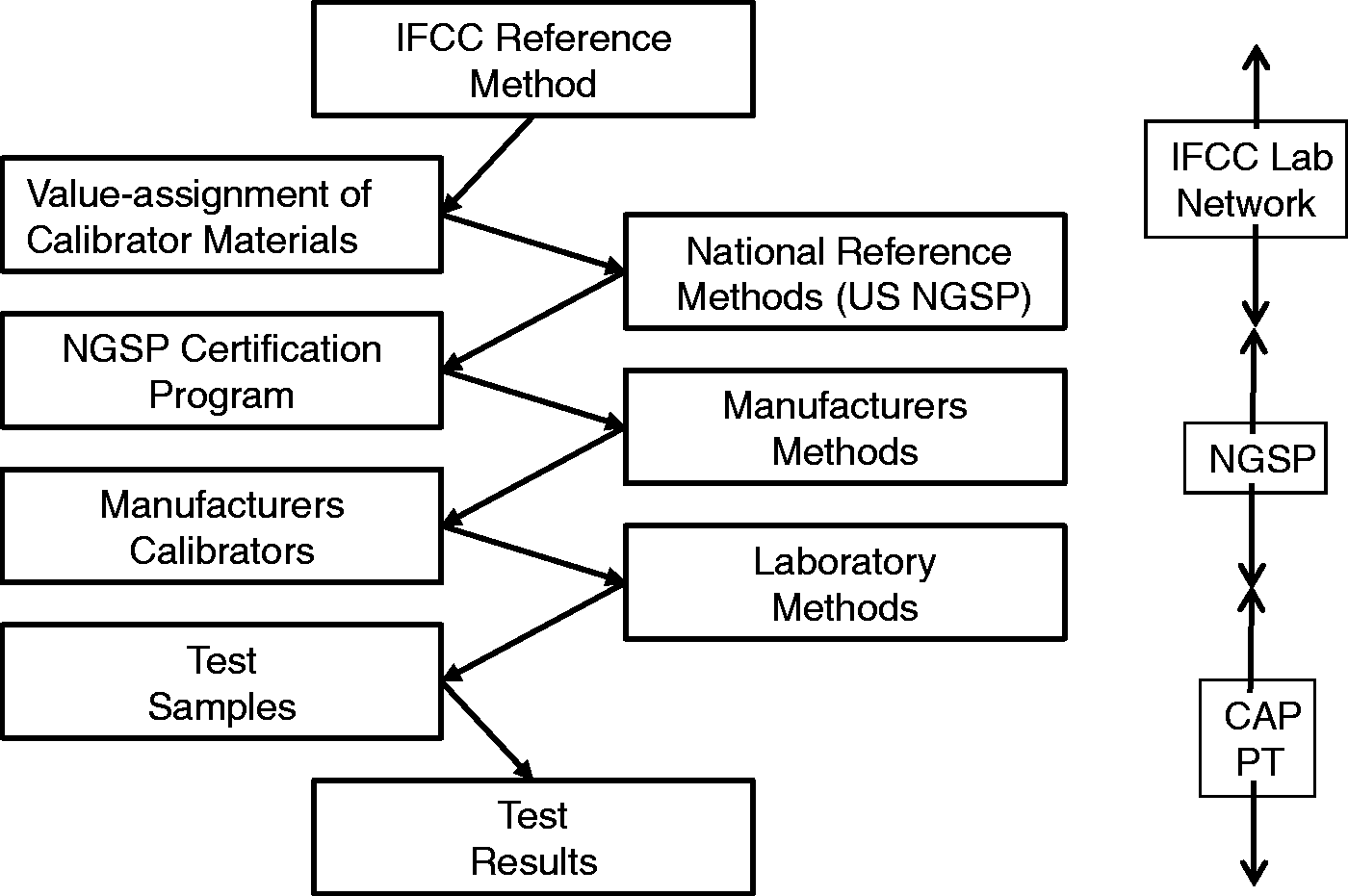
Nonetheless, significant biases still exist between method subgroups, as illustrated in Figure 5 which shows the variation among 26 subgroups in a CAP survey from 2014.
26
The quality requirement of 6% is shown by the band in the middle. Performance of different method subgroups is shown by the vertical lines where the midpoints represent the average result for each subgroup and the confidence interval represent the distribution of results from different laboratories using that same method. The average absolute bias is 2.3% and the maximum observed bias is 6.0%. Given the goal of 6% maximum error, bias on average consumes 38% of the error budget and in eight method subgroups consumes over 50% of the error budget.
Comparability of HbA1c results for 26 method subgroups in a 2014 survey by the College of American Pathologists. Assigned value is 6.49 %Hb. Data obtained from NGSP website, www.ngsp.org.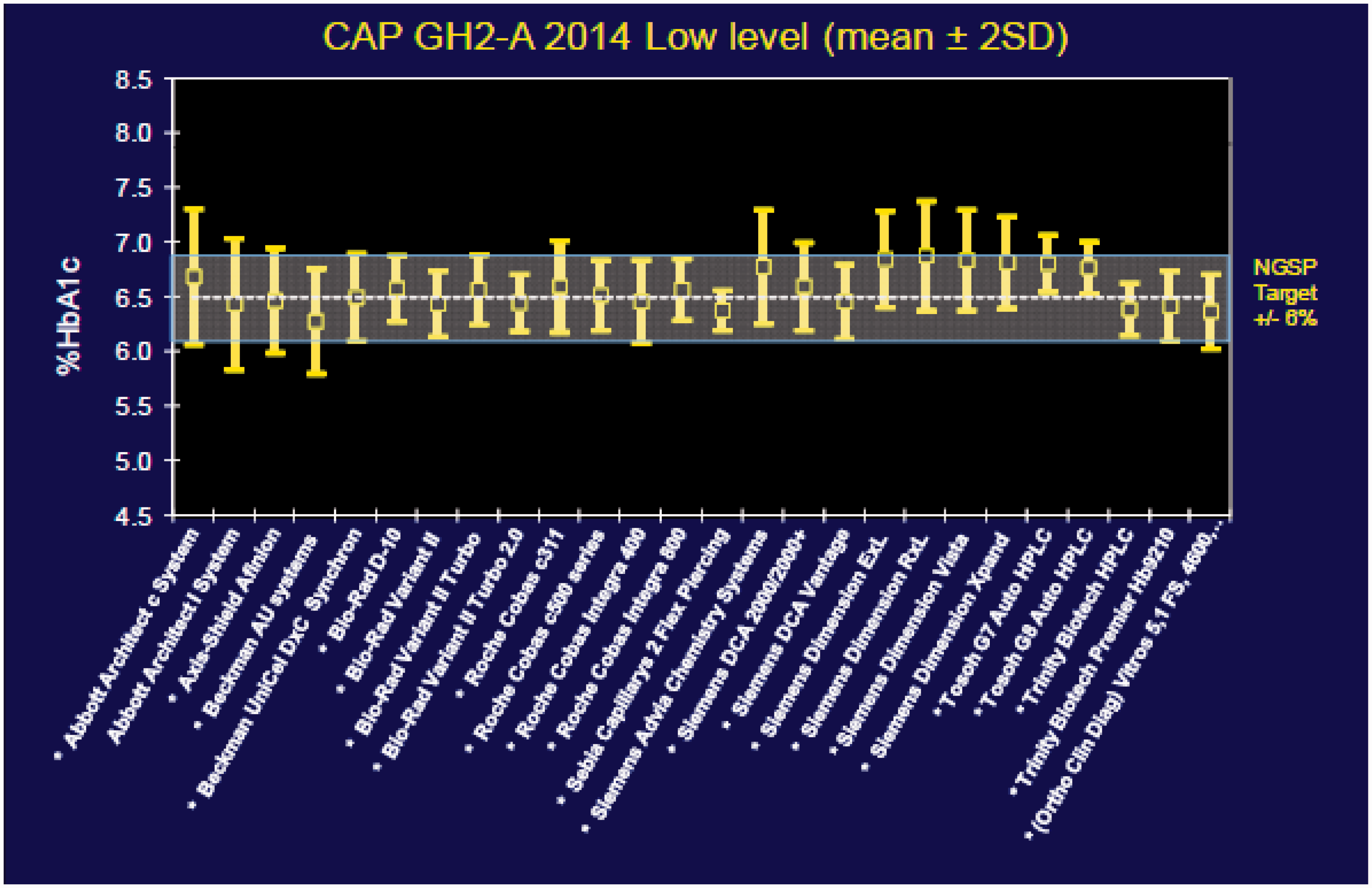
These survey results for HbA1c show that achieving comparability of results across methods, across laboratories, across health networks and systems and across geographic borders is still a very difficult task. For many other biologic measurands, the traceability infrastructure simply does not exist. Traceability is a goal, but harmonization of results with a specified calibrator or selected examination procedure is a more immediate and practical approach. 27 That means settling for the ‘relative truth’ and consistency of results is a more practical, albeit less than ideal, strategy in medical laboratories today.
In practice, medical laboratories are dependent on manufacturers for traceability and harmonization. But, they still must monitor the comparability of results through EQA or PT programmes and should actually utilize the results of such programmes in making purchase decisions for new analytic systems.
Step 3. Validate safety characteristics
According to ISO guidance on assessing the risk of test systems and devices, 28 manufacturers should consider that the performance characteristics that affect the accuracy of an examination are ‘safety characteristics.’ Patient safety will be affected by the hazardous conditions that result from failure to achieve the performance required for the intended use. Validation of examination procedures is therefore a critical part of risk assessment for patient safety. In the laboratory, verification of a manufacturer's performance claims is supposed to ensure patient safety. However, this assumes that the manufacturer correctly established its performance claims and that those claims have been validated against requirements for intended use, even though manufacturers typically compare performance to a predicate device, often the previous generation of their own examination procedure, rather than comparing to reference methods and materials. Therefore, laboratories should validate new methods against their own defined requirements for intended use, rather than just verify a manufacturer's claims for performance.
Standard validation protocols are available from CLSI29,30 and are commonly referenced in published validation studies. However, the analysis and interpretation of the experimental data seldom consider the quality required for intended use. A simple way to do this is to calculate a Sigma-metric or use a graphical tool such as the Method Decision Chart to evaluate quality on the Sigma-scale. 31 Knowledge of quality on the Sigma-scale provides key insight into the needs for quality control because the metric encompasses the quality required for the examination and the precision and accuracy observed for the examination procedure.
A Sigma-metric is typically calculated as follows:
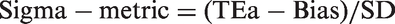
HbA1c provides a practical application. A recent study by Lenters-Westra and Slingerland
33
provided experimental results for precision following the CLSI EP05 protocol
29
and results for accuracy by comparison with reference methods following the CLSI EP09 protocol.
30
The authors compared 7 POC devices to three different ‘secondary reference measurement procedures’ that were available in the European Reference Laboratory for Glycohemoglobin at Zwolle in The Netherlands. The results are summarized by the Method Decision Chart shown in Figure 6. Two of the devices provide less than 2-sigma quality, two others provide less than 3-sigma quality, one overlaps the 3-sigma line, and two overlap the 4-sigma line. Keep in mind that all of these methods are certified by NGSP as providing equivalent quality results based on patient comparative results submitted by the manufacturer but there still are significant biases between method subgroups.
Method Decision Chart for HbA1c with Tea = 6.0%. Diagonal lines from top to bottom represent 2σ, 3σ, 4σ, 5σ and 6σ. Vertical lines each represent one of the seven POC devices tested. The x-coordinate of the vertical line represents the precision observed for the POC device. The y-coordinates, shown by the points on the vertical line, represent the bias when compared to each of the three ‘secondary reference measurement procedures’ used in the comparison of methods study.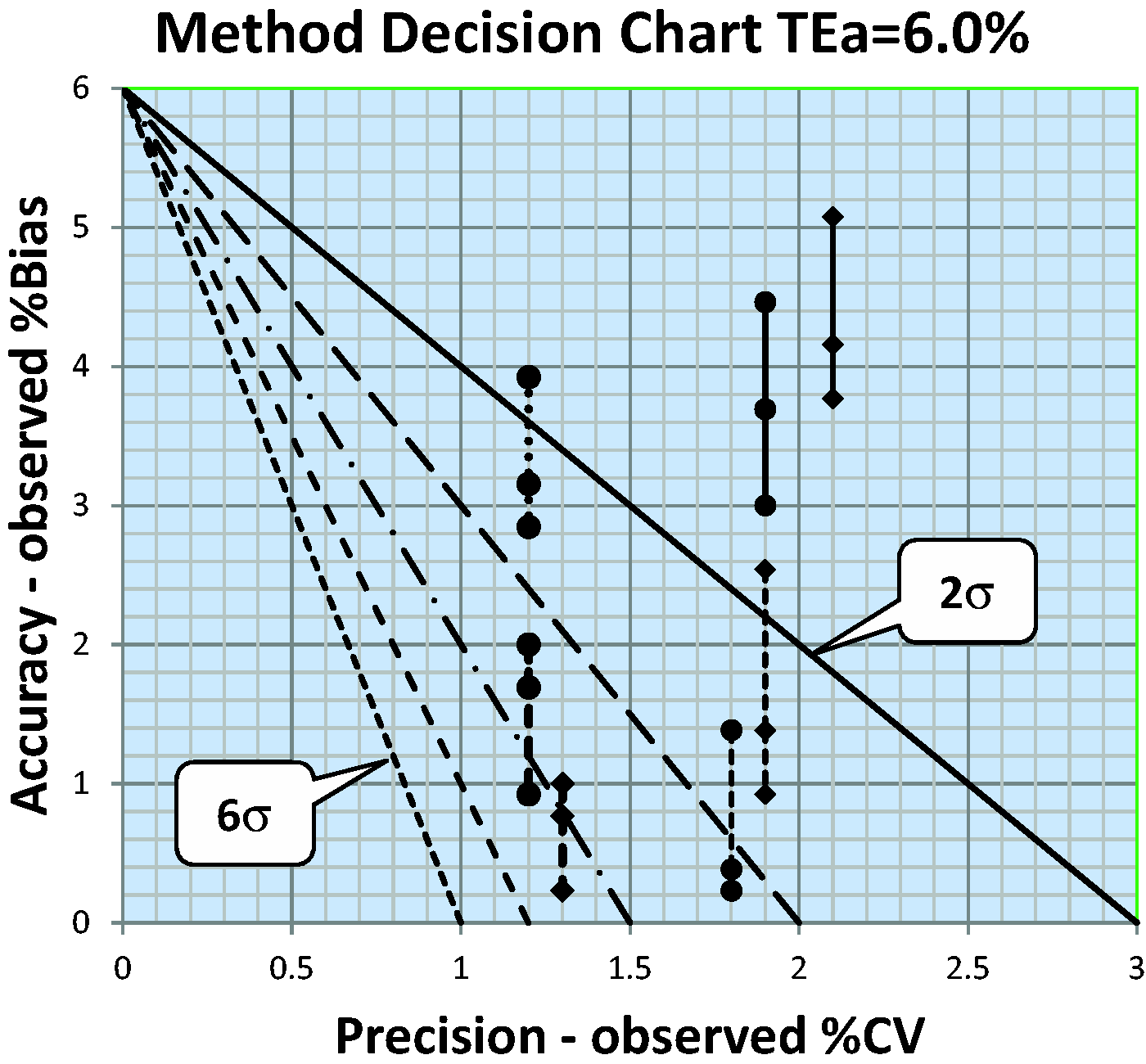
Our point here is that method validation and acceptable method performance are prerequisite to the design and implementation of SQC procedures. If performance is NOT acceptable under stable operating conditions, no amount of QC can change or improve that performance. QC can only monitor performance, and when properly designed, alert analysts to the presence of additional errors that occur because of unstable performance.
Step 4. Implement the analytic system
An SOP must be developed to formalize the steps for operating the new system. Analysts must be trained to follow the SOP. Manufacturers support the initial training for a new analytic system, but laboratories should also develop their own in-service training programme to ensure adequate training for new analysts over the life-cycle of the analytic system. In-service training in basic QC practices is an on-going need in many laboratories.
It is also critical to review or implement requirements for pre-examination and postexamination processes. Requirements for intended use should be specified and their implementation reviewed through audit of appropriate documents. During this review, the laboratory should also begin identifying controls that will be needed to ensure the quality of the pre-examination and postexaminations phases of the process. For example, specimen and sample acceptability criteria should be established and controls included in the laboratory QC Plan. The laboratory should also survey its capabilities for implementing different kinds of control mechanisms for the analytic phase. The CLSI EP23A document 34 describes various controls that may be included in a laboratory QC toolbox for developing QC Plans on the basis of risk management.
Step 5. Formulate a total QC strategy
The amount of QC that is needed depends on the performance of the examination procedure relative to the quality required for intended use. When the quality required for intended use is defined by TEa and the quality observed for an examination procedure is described in terms of bias and imprecision, then the Sigma-metric provides a comparison of the quality needed to the performance observed. This Sigma-metric is a ‘process capability index’ whose use is well established in industrial process management. For example, the industrial process capability index Cpk is related to the Sigma-metric as follows:
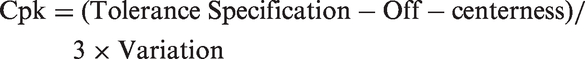
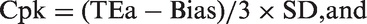
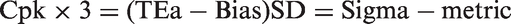
For industrial production, the minimum Cpk that is acceptable is specified to be 1.0, which corresponds to 3-sigma. The ideal or desirable Cpk is 2.0, which corresponds to 6-sigma.
Given that process capability indices have been used for many years in manufacturing industries and considering that a medical laboratory is a production operation, the use of a Sigma-metric as a process capability index provides an established approach for managing the laboratory testing processes. However, there is a need for guidance on how to use measures of Sigma-quality for management purposes.35,36
The initial guidance relates to a general strategy where the total QC plan is optimized on the basis of the observed sigma-quality.
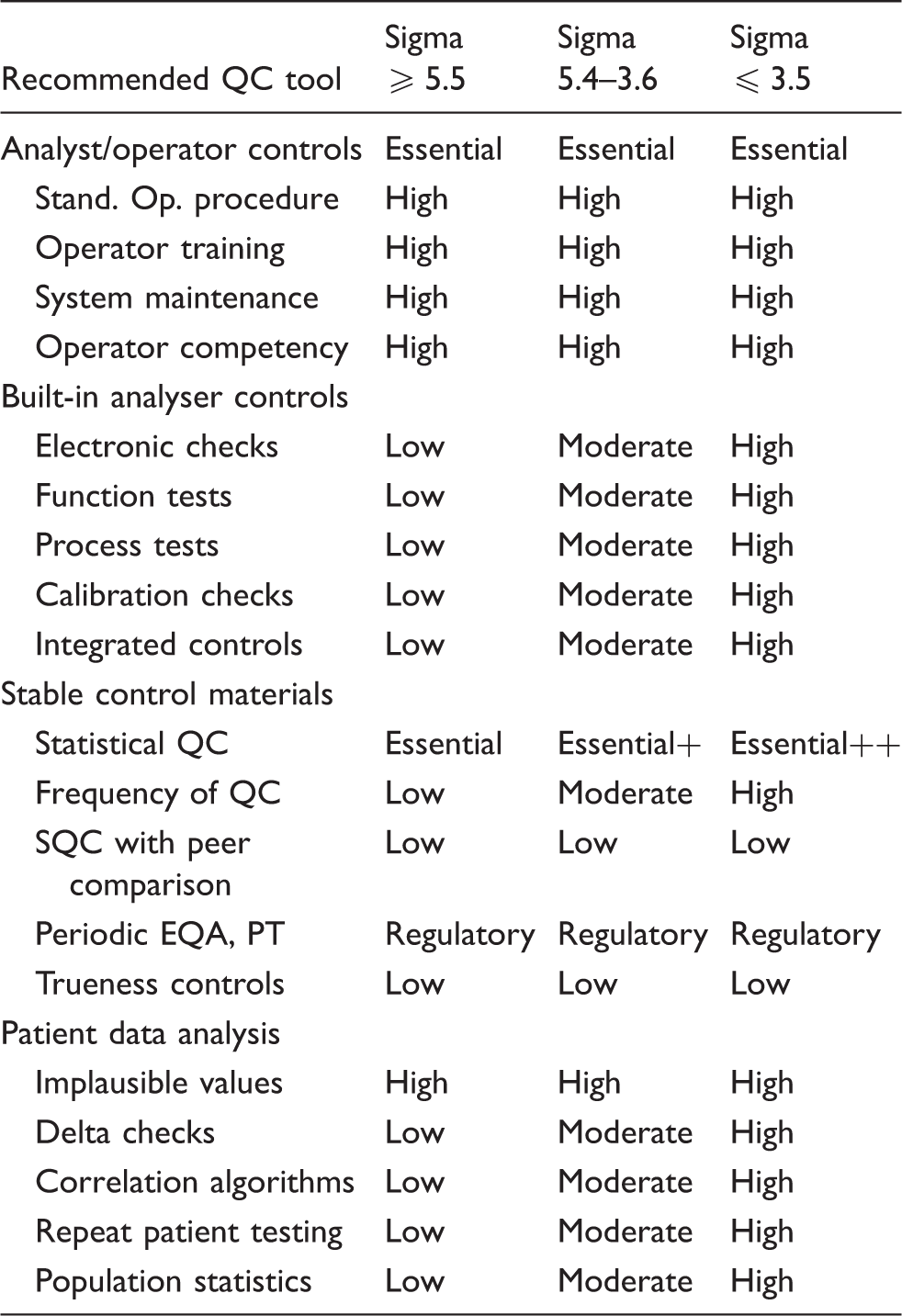
A high sigma TQC strategy is appropriate for ≥5.5σ and should emphasize the ‘right-sizing’ of SQC procedures to ensure detection of medically important errors. The laboratory should follow the manufacturer's QC instructions and other good laboratory QC practices and should add preanalytic and postanalytic controls to monitor the total testing process. A moderate sigma TQC strategy is applicable for 5.4 to 3.6σ and requires more SQC, typically doubling the number of control measurements per run and employing multirule criteria. The laboratory should aggressively follow the manufacturer's QC instructions and consider additional controls for the analytic process, as well as controls for preanalytic and postanalytic phases of the total testing process. For sigmas of 3.5 or less, the maximum affordable SQC is needed but even that may not be adequate for detecting medically important errors; therefore, it will be necessary to add other analytic controls to ensure the quality of examinations. Here is where a risk assessment is useful to identify possible failure modes and introduce additional controls to mitigate their effects, as well as identifying preanalytic and postanalytic controls. Patient data QC procedures may be helpful. Analyst training and experience will be important for providing eyes and ears that are sensitive to changes and variations in the process.
Sigma priorities for inclusion of controls in total quality control plan.
Step 6. Select or design a statistical QC procedure
A recommendation for planning SQC procedures was a major focus of the 2003 QC review. 1 The key information that is needed is the probability of rejecting an analytical run as function of the size of the errors occurring in a run. Different control rules and different numbers of control measurements have different ‘power curves’; therefore, a graphical display of the probability of rejection on the y-axis vs. the size of systematic error on the x-axis is useful for comparing the rejection characteristics of different SQC procedures.
Figure 7 shows an example that compares the power curves for several single and multirule QC procedures that are identified in the key at the right. The curves top to bottom correspond to the rules in the key top to bottom. The probability for rejection (P) of an analytic run is plotted on the y-axis vs. the size of the systematic error on the bottom x-axis and the Sigma-metric on the top x-axis. The condition where the only error present is the stable random error of the measurement procedure is shown by the y-intercept of each power curve and identifies the probability for false rejection (Pfr). The design goal is to keep Pfr as low as possible while selecting a QC procedure that provides a probability of error detection (Ped) of 0.90, or 90% chance of detecting a medically important error. The desired error detection is illustrated by the dashed line that intersects the power curves at 0.9 on the y-axis.
Sigma SQC Selection Tool for two levels of controls. The probability for rejection is plotted on y-axis vs. the size of systematic error on bottom x-axis and the sigma-metric on the top x-axis. In the key at the right, the different power curves correspond, top to bottom, to the list of control rules, the probability for false rejection (Pfr), total number of control rules (N), and number of runs (R) over which the rules are applied. This chart was produced by the EZ Rules3 computer program.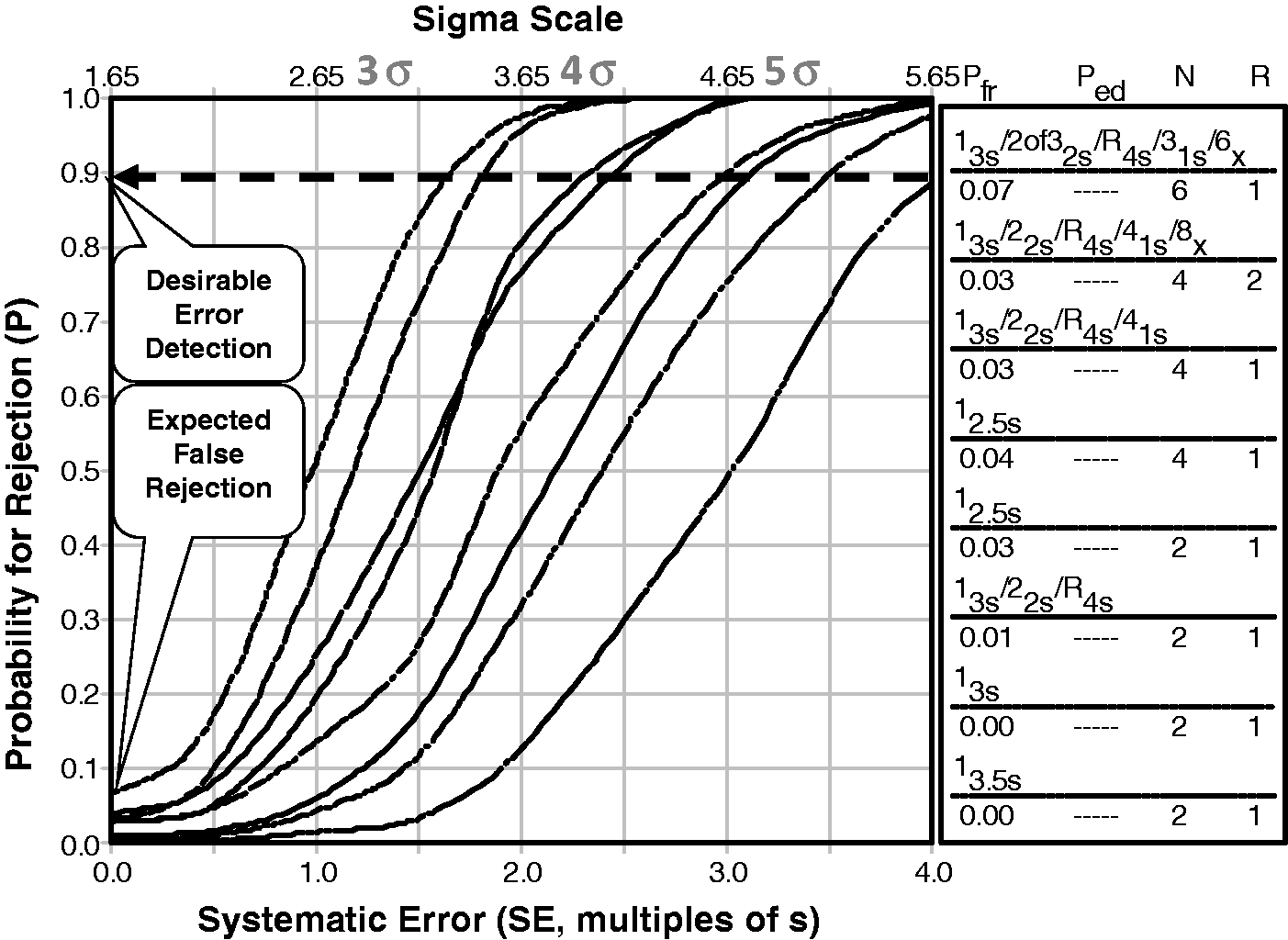
The systematic error that is shown on the x-axis is a multiple of the SD of the method, e.g. a value of 2.0 indicates a systematic shift or bias that is equivalent to two times the value of the SD; a value of 3.0 means three times the value of the SD, etc. The critical systematic error (ΔSEcrit) is calculated as follows:
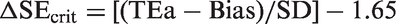
This ‘critical error graph’ has been adapted to include a Sigma-scale to further simplify applications. Note that the critical error equation can be arranged as follows:
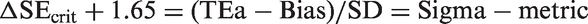
Therefore, a Sigma-scale can be imposed by adding 1.65 to the ΔSEcrit value, as shown by the x-axis scale at the top of Figure 7. The selection of SQC procedures can be made directly by locating the value of the Sigma-metric on the x-axis, then dropping a vertical line to intersect the power curves. This refined graph is called a ‘Sigma-metric SQC Selection Tool’ and was included in the 2006 edition of the CLSI C24A3 guideline for Statistical QC.
37
Figure 7 is intended for use with two levels of controls, whereas Figure 8 is intended for three levels of controls.
Sigma SQC Selection Tool for three levels of controls. The probability for rejection is plotted on y-axis versus the size of systematic error on bottom x-axis and the Sigma-metric on the top x-axis. The different power curves correspond, top to bottom, to the list of control rules and N in the key at the right side.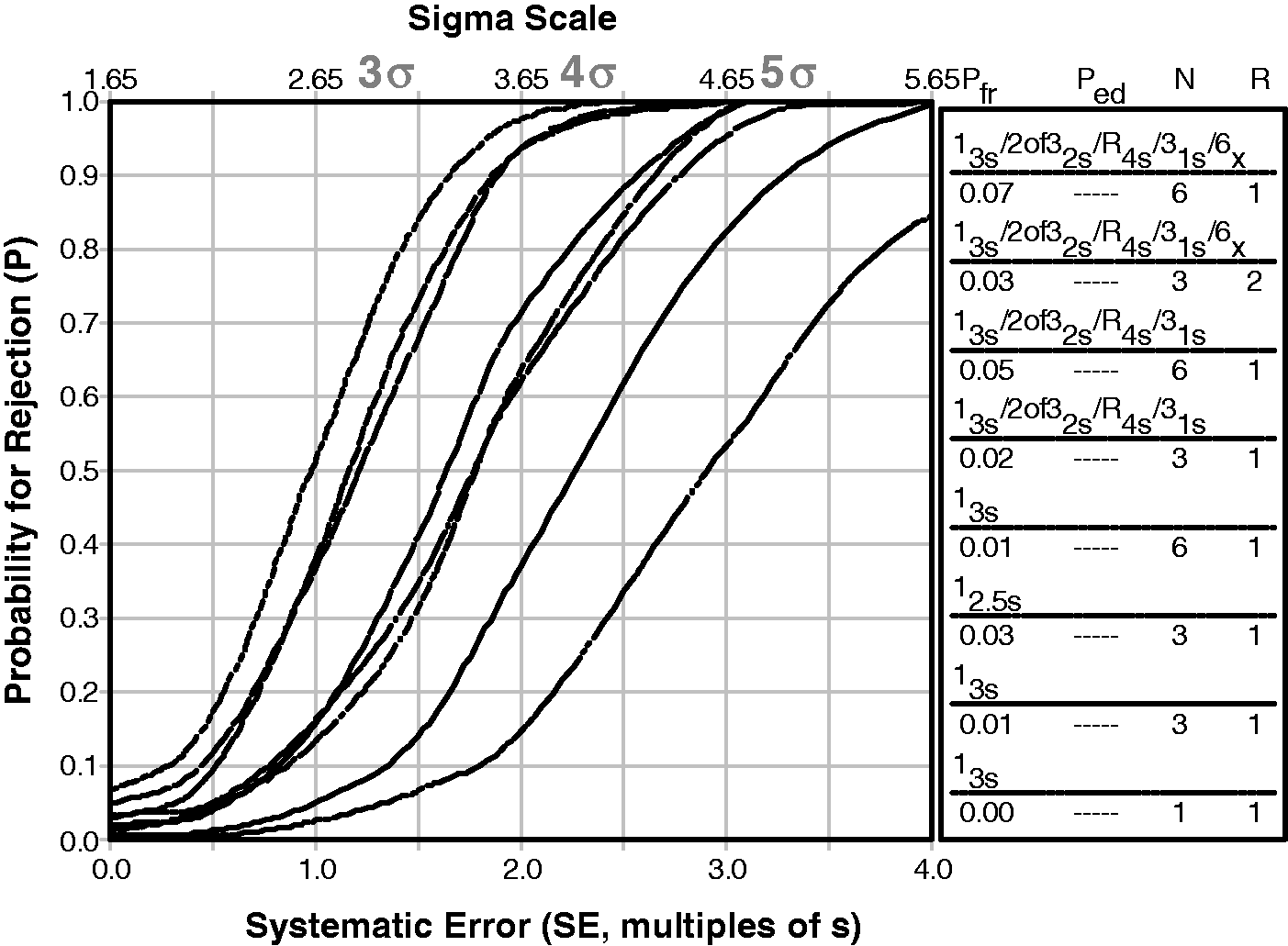
The 2003 review
1
also described the use of a Chart of Operating Specifications, or OPSpecs chart. In many applications, the OPSpecs chart may be preferred because its format is similar to the Method Decision Chart. However, many analysts find the OPSpecs chart initially difficult to understand even though once mastered, it is easy to use. In our continuing efforts to simplify the selection of appropriate SQC procedures, we have recently introduced a new graphic tool called ‘Westgard Sigma Rules’.
8
This graphic provides the traditional flowchart for the application and interpretation of Westgard Rules, but includes a Sigma-scale at the bottom, as shown in Figure 9. To select the SQC procedure appropriate for an observed Sigma-metric, locate the value of sigma on the scale at the bottom, follow the arrow up, identify the control rules to the left side of the arrow and finally identify the total number of control measurements and number of runs from the values given for N and R, respectively.
‘Westgard Sigma Rules’ Graphic Tool for three levels of controls. Locate sigma-metric on scale at the bottom, read up and identify the control rules, number of control measurements (N), and number of runs (R) to the left of the arrow. For example, if the sigma-metric is five, then the appropriate SQC procedure is a 13s/22s/R4s with two control measurements in a single run.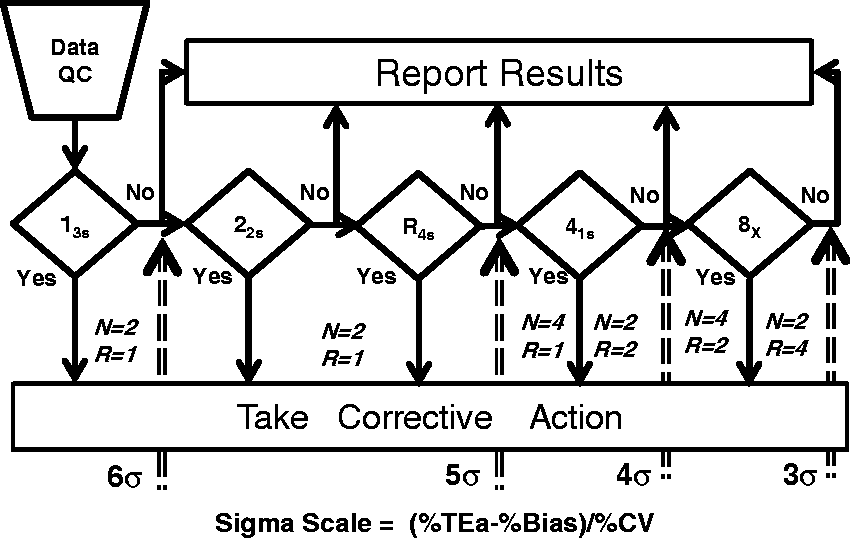
Here are some examples for HbA1c.
TEa = 6.0%, Bias = 1.0%, CV = 1.0%, Sigma-metric = (6.0−1.0)/1.0 = 5. Use a 13s/22s/R4s multirule procedure with N = 2 and R = 1. TEa = 6.0%, Bias = 0.0%, CV = 1.5, Sigma-metric = (6.0−0.0)/1.5 = 4.0. Use a 13s/22s/R4s/41s multirule procedure with N = 4 and R = 1, or with N = 2 and R = 2. TEa = 6.0%, Bias1.5%, CV = 1.5%, Sigma-metric = (6.0−1.5)/1.5 = 3.0. Use a 13s/22s/R4s/41s/8x multirule procedure with N = 4 and R = 2, or with N = 2 and R = 4.
Figure 10 provides a similar graphic for ‘Westgard Sigma Rules’ that are appropriate for three levels of controls.
8
‘Westgard Sigma Rules’ for three levels of controls.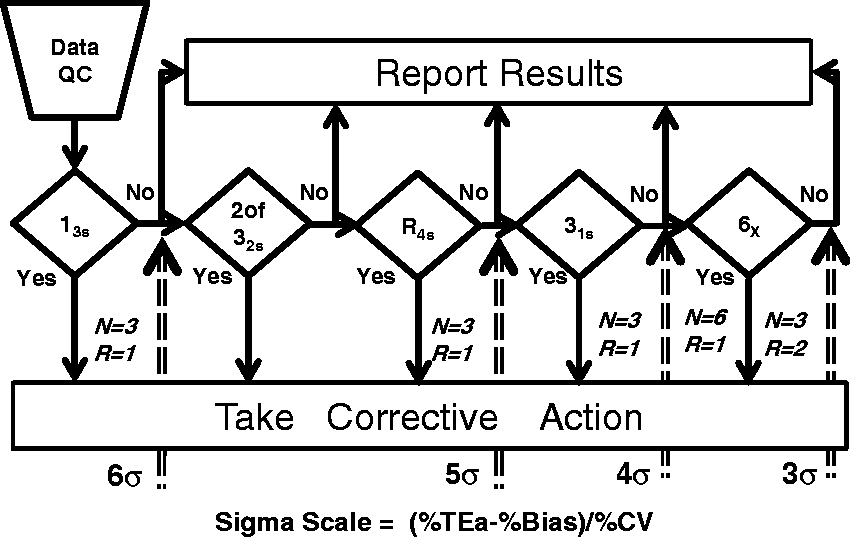
Other approaches for selecting and designing SQC procedures have also been recommended in the recent literature. Woodworth et al. 38 described a risk assessment methodology that utilized a patient-weighted Sigma-metric 39 as the key factor for characterizing the quality of measurement procedures, estimating the risk from reporting unreliable patient results and selecting appropriate SQC procedures. The mathematical and statistical calculations are considerably more sophisticated and require computer support; nonetheless, the sigma quality of the measurement procedure is the key risk factor and the most important parameter for selecting an appropriate SQC procedure.
For the seven HbA1c methods studied by Woodworth et al., 38 the estimates of the Sigma-metrics were 0.36, 1.43, 1.57, 2.29, 2.36, 2.84 and 3.90 when TEa was 6.0%. All of these examination procedures were certified as equivalent by NGSP and were in routine operation in central laboratories. In recommending SQC procedures, the authors stated that those examination procedures where sigma was less than 3 needed the maximum QC of three levels three times per day. By comparison, these examination procedures fall off the end of the Sigma-scale on the ‘Westgard Sigma Rule’ graphics, thus we would also make a recommendation to perform the maximum amount of QC that is affordable. For the best performing method that has a Sigma-metric near 4, the appropriate SQC would be a 13s/22s/R4s/41s multirule procedure with 4 control measurements per run.
Another approach that was recently recommended by Ceriotti et al. 40 is the use of an ‘acceptance control chart’ where the measurement uncertainty is first characterized, then an acceptance zone established by subtracting the expanded measurement uncertainty from the TEa limits for the maximum permissible errors. The authors state that optimal control is achieved when the relationship between the maximum permissible error (TEa) and the expanded measurement uncertainty (U, 95%) is 2.5 and that when TEa/U is close to 1, it will be impossible to control the measurement procedure. Given that U represents a 2SD range, a TEa/U ratio of 2.5 corresponds to a 5-sigma process and a ratio of 1.0 corresponds to a sigma of 2. Similar conclusions on controllability can be reached by calculating Sigma-metrics; in addition, more specific guidance on the selection of control rules and the number of control measurements is provided using the Sigma-related SQC planning tools.
Step 7. Develop a total QC plan
Although ISO 15189 has had only minor impact on quality practices in US laboratories, ISO 1497128 is having major impact on the use of risk-based QC Plans, which have become an option for compliance with the US CLIA requirements for QC. That option is called an ‘Individualized Quality Control Plan’, or IQCP, 41 and is described as a combination of three parts: a risk assessment to identify sources of errors; a control plan to identify mechanisms for mitigating those risks; and a quality assessment programme to monitor performance and identify needs for improvement.
In principle, a QC plan is a good idea for bringing together the many activities that are necessary for ensuring the quality of an examination procedure, including the pre- and postexamination parts of the total examination process. Risk assessment, however, is a new and difficult activity for most medical laboratories because they have little or no training or experience in doing this. CLSI has published a guideline for ‘Laboratory Quality Control Based on Risk Management, 34 but the assessment of risk is qualitative and subjective, the process is complicated and standardization is needed to provide reproducible and consistent results. Risk assessment can be made more quantitative and objective by application of Six Sigma concepts and metrics. 42 However, the current CMS/CDC guide 41 only emphasizes the identification of hazards without actually assessing the risk of those hazards and does not even adhere to the CLSI guidance. In short, IQCP is a risk-based QC plan without risk assessment, which is an obvious shortcoming and limitation that should be addressed for this approach to be valid.
Meanwhile, laboratories can take a step in the right direction by developing what we call a ‘total QC plan,’ which optimizes SQC to detect medically important errors, then includes additional controls to monitor possible failure modes in the pre- and postexamination parts of the total examination process. Risk assessment can be used to identify those failure modes, but we can start with qualitative models, knowing that analytical errors are being monitored quantitatively by right-sized SQC procedures and that the need for other analytical controls can be prioritized for the observed Sigma-metric (see Table 1). In this context, risk-based controls become a logical extension of a total QC Plan that includes a right-sized SQC procedure.
Outline of information that may be included in a laboratory total quality control plan.
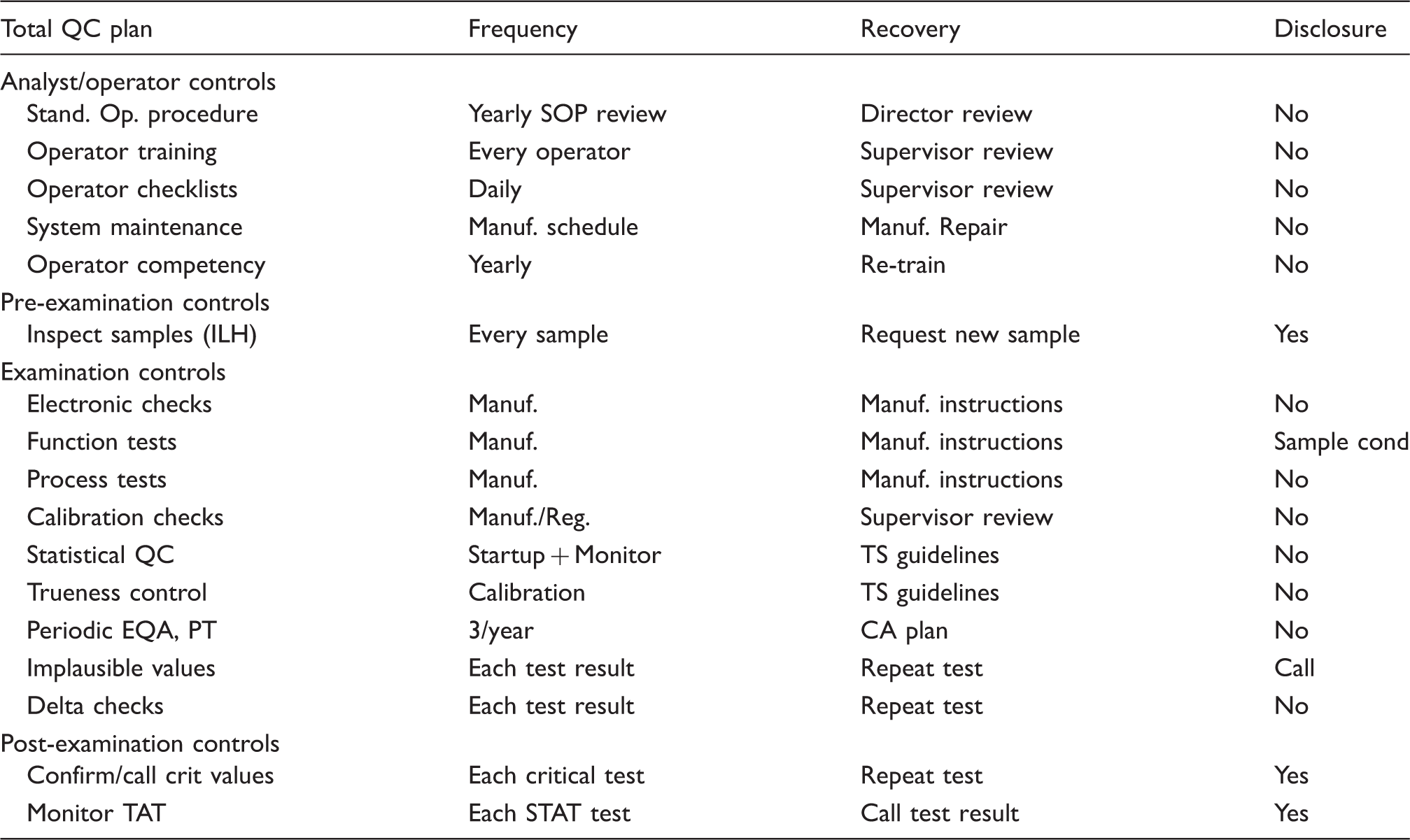
Step 8. Implement the QC plan
A wide variety of control mechanisms are recommended in the CLSI EP23A QC toolbox, 34 but controls are not all created equal! The mere existence of a control does not mean it will accomplish its intended purpose. In particular, the error detection capabilities are seldom known, except for SQC procedures, which is why SQC is needed to verify the attainment of the intended quality of results.
Nonetheless, there is an increasing interest in the use of patient data for QC, including recommendations for moving averages and moving medians as a new paradigm for QC.43,44 The popularity of related algorithms, such as the Average of Normals (AoN),45–47 has waxed and waned since their introduction in the 1960s. In the past half century, patient data controls have yet to be proven practical for many production laboratories, regardless of the massive increase in available informatics and the widespread interest in applications.
In general, patient data control mechanisms should be considered as a complement to SQC with stable control materials, not a substitute. For population distribution algorithms, a certain number of patient results must be accumulated before there can be any assessment of control. Other patient data controls, such as delta checks, work only for individual patient results for which a recent past result is available for comparison. Likewise, limit checks for widely variant results are useful for individual patient results, but they do not help provide any general assessment of the control status of a run. The use of stable control materials and SQC still provides the best primary control mechanism and may be supplemented, but not replaced, by patient data controls.
In small laboratories where few patient samples are analysed and it is difficult to analyse stable control materials, repeat patient tests might provide a useful control for monitoring variability. All operators recognize that they should be able to get the same test result if they analyse the same patient again, thus repeat patient tests will help even unskilled operators to understand that test results may vary due to the testing process itself.
One situation where there is a strong recommendation for the use of patient samples is for comparison of performance when there are changes in reagent lot. Miller et al. 48 strongly recommend that a group of 10 or more patients be tested by both the old reagent lot and the new reagent lot because control materials may not be commutable, i.e. may not behave the same as patient samples. CLSI 49 has developed a detailed, though complicated, planning process for determining the optimal number of patient samples that should be used, taking into account the Critical Difference in results defined by the laboratory (i.e. the average difference between reported results from a set of patient samples that corresponds to the limit a laboratory is willing to accept), the statistical power desired and the performance of the examination procedure relative to the Critical Difference.
Step 9. Verify the attainment of the intended quality of test results
Given a properly designed SQC process that will detect medically important errors, one of the ongoing issues in routine production is the frequency of controls. Some initial guidance is provided by the Westgard Sigma Rules in terms of total number of controls per run (N) and the number of runs over which controls are analysed (R). For example, the four control measurements required by a multirule SQC procedure might be spread over two 4-h runs (N = 2, R = 2), rather than an 8-h run (N = 4, R = 1). For small numbers of patient samples, a single run would be more efficient. For large numbers of patient samples, scheduling the workload over two runs may be more effective.
The frequency of QC should also be adapted for ‘events’ or changes in the testing process that need to be qualified by analysis of new controls, as recommended by Parvin. 50 There are both ‘expected events’ and ‘unexpected events.’ The first refers to known, scheduled, or observed changes that occur at specific times; the latter refers to unexpected changes that might occur anytime. An important strategy is to schedule controls for all expected events, such as changes in reagent lots, calibrator lots, system maintenance, replacement of parts, changes in environmental conditions and possibly changes in analysts or operators. For unexpected events, it is important to periodically monitor the testing process to limit the possible exposure to unknown changes that may affect the quality of patient test results.
The SQC design for expected events should include the right control rules and right number of control measurements to detect medically important errors, e.g. right-sized Westgard Sigma Rules. For unexpected events, a monitoring design should be employed that uses a single control rule and periodic controls spaced throughout the analytic run. One practical consideration is the number of patient samples that would need to be retested if a run were determined to be out-of-control. The cost of repeat testing should be weighed against the cost of analysing periodic controls throughout the run.
For example, the laboratory might employ a 13s/22s/R4s/41s multirule design with four control measurements whenever scheduled changes occur, such as the change of reagents and analysts in the daily start-up of a test system. For a batch operation, the controls may be spaced throughout the run and evaluated before release of the batch of test results. For continuous reporting operation, ‘start-up controls’ may be located at the beginning of the run in order to quickly assess the control status of routine testing, then additional individual ‘monitor controls’ should be spaced throughout the run and evaluated using a 13s or 12.5s control rule at reasonable intervals for reporting of patient test results.
Determining the optimal frequency of controls is currently an unresolved issue. More sophisticated models are being developed by Parvin and Yundt-Pacheco. 51 A practical application is illustrated for HbA1c examination procedures in the paper by Woodworth et al. 38 The models are mathematically complex and would only be practical with informatics support. However, it is interesting to note that the recommendations for appropriate SQC procedures made by Woodworth make reference to a table of ‘Sigma values with their optimal Westgard (multi) rules’ published by Schoenmarkers et al. 52 and derived from Charts of Operating Specifications.
Meanwhile, until better guidance and tools are available to establish frequency of controls, medical laboratories should consider their regulatory requirements as setting the minimum frequency of controls. To that, the laboratory should add controls for expected events to evaluate known changes in the testing process and also add controls for unexpected events to monitor the process during routine operation and minimize the risk and costs of unexpected changes. Finally, risk-based controls should be added to monitor specific failure modes of the particular test and test system.
There are many other practical issues in routine SQC applications and operations, as discussed recently by Kinns et al. 53 These authors provide a lot of practical advice that is particularly oriented to NHS laboratories, including guidance for selection of SQC materials, assigning an SQC range, design of an SQC system on the basis of the observed Sigma-metric, frequency of SQC, use of patient data, practicality and cost of implementation, review of SQC data and applications within a laboratory network. The troubleshooting diagram, adapted from Schoenmarkers et al., 52 is particularly useful for identifying problems with SQC materials, reagents, calibrators, analysers and the environment. Readers should consider the papers by Schoemarkers and Kinns as essential background for application and operation of SQC procedures.
Step 10. Measure quality and performance
With implementation of a scientific quality control process, as described in steps 1 through 9, it is important to monitor quality and performance of the production process to ensure that quality goals are being achieved in routine operation. Of particular interest here are the determination of measurement uncertainty (MU) from SQC data and the determination of bias from PT and EQA data.
In the earlier 2007 edition of ISO 15189, the determination of MU was recommended ‘when relevant and possible.’ ‘Relevant’ led to discussion of whether or not physicians needed this information for their use and interpretation of test results. ‘Possible’ often invoked arguments that the GUM methodology
54
for estimating MU was too complicated to be practical in medical laboratories. However, the 2012 document
2
removes those qualifications and makes the determination of MU a requirement: 5.5.1.4 The laboratory shall determine measurement uncertainty for each measurement procedure in the examination phase used to report measured quantity values on patient samples. The laboratory shall define the performance requirements for the measurement uncertainty of each measurement procedure and regularly review estimates of measurement uncertainty.
A practical consideration is the number of control measurements that should be included to obtain a reliable estimate of MU. While there is a ‘rule of thumb’ that a minimum of 20 control measurements should be used to calculate an SD for setting control limits, many more are needed to obtain a reliable estimate of the SD. For example, if we assume a true standard deviation of 10 units, the 90% confidence interval will range from 7.4 to 15.9 when N = 20, i.e. an SD as low as 7.4 could be observed, which is 26% low, or an SD as high as 15.9 could be observed, which is 59% high. For N = 100, the confidence interval is 9.0 to 11.3, i.e. the reliability of the estimate of the SD is much better, within approximately 10% of the correct value. Therefore, it would seem prudent to aim for at least 100 measurements when estimating MU and those measurements should be collected over a period of three to five months in order to satisfy the requirement for intermediate precision conditions. That makes the determination of MU impractical for the initial validation of an examination procedure but appropriate for the on-going monitoring of quality and performance.
Another issue with MU has been the consideration of bias. 55 Clinical laboratory scientists have commonly determined the analytic total error as a measure of quality and performance. However, metrologists argue it is wrong combine imprecision and bias; instead, bias should be eliminated or corrected so it need not be considered in determination of MU. Because metrologists have such a strong influence in ISO, total error is discouraged.
Nonetheless, the estimation of bias is essential for managing the quality of examinations in medical laboratories. PT and EQA data show that bias is a real problem and has not been eliminated nor corrected even in global standardization programmes, such as the IFCC programme for global standardization of HbA1c. ISO 15189 requires laboratories to participate in PT and EQA programmes, whose main value is to estimate any bias between the results from an individual laboratory and an assigned target value. To account for bias along with MU, we have recommended the assessment of quality on the Sigma-scale and demonstrated the implications for SQC practices. 56 More recently, we have proposed that PT and EQA programmes adopt a ‘Sigma Proficiency Assessment Chart’ 57 to display both the bias and imprecision of examination subgroups and better understand the quality of current examination procedures.
An example is shown in Figure 11 for HbA1c results in a 2014 CAP survey sample whose concentration is 6.49 %Hb (47.4 mmol/mol), which is very close to the diagnostic cut point of 6.5 %Hb (47.5 mmol/mol). The ‘>’ shaped lines delineate the borders for 2-sigma and 3-sigma quality. For example, the points to the right of the 2-sigma line represent examination subgroups whose quality is less than 2-sigma. The points between the two lines represent examination subgroups better than 2-sigma but still not up to the industrial standard of 3-sigma for acceptable production processes. Only one examination subgroup shows better than 3-sigma quality. Note that this is the same data shown earlier in Figure 5.
Sigma Proficiency Assessment Chart for 2014 College of American Pathologists (CAP) survey results for HbA1c GH2-01 sample with concentration of 6.49%Hb. Tea = 6.0%. Each point represents the observed bias (y-axis) and the observed imprecision (x-axis) for one of 26 examination subgroups. Results represent a total of 3187 laboratories. Note these are the same data shown in Figure 7.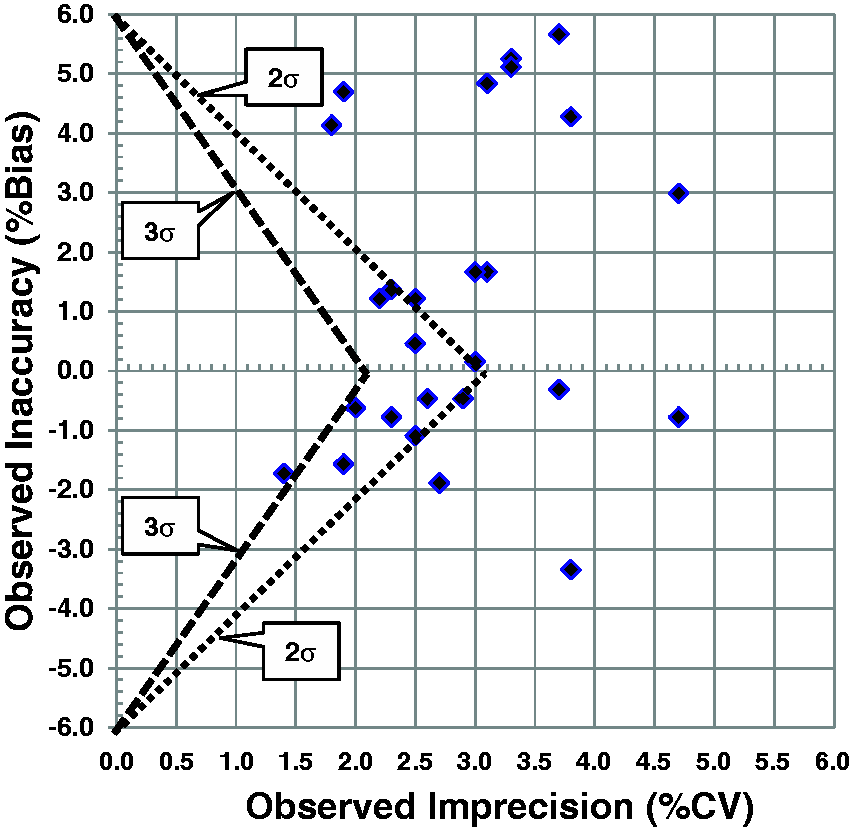
Weykamp et al. 58 have also recommended adoption of a Six Sigma model for evaluating quality targets and have demonstrated the quality of current methods using PT/EQA data and a graphic summary similar to the Method Decision Chart discussed earlier. Interestingly, they considered that the Sigma-metric represented the risk of poor quality results and recommended that routine laboratories should achieve at least 2-sigma quality and that laboratories participating in clinical trials should achieve 4-sigma quality. They also used data from a 2014 CAP survey to illustrate the quality achieved by 26 different manufacturers or instruments when TEa was set at 5 mmol/mol (which corresponds to 4.6% TEa in %Hb or NGSP units). Only 12 of the 26 instrument groups met their recommended 2-sigma criterion. They also demonstrated how the Six Sigma model could be used for assessing results from individual laboratories when both bias and within lab imprecision figures were available.
Finally, remember that ‘the laboratory shall define the performance requirements for the measurement uncertainty of each measurement procedure and regularly review estimates of measurement uncertainty’. 2 A practical approach may be to employ a TEa quality goal and prepare a normalized Method Decision Chart, which allows a graphical presentation of both bias and imprecision, without any need to mathematically combine the estimates, thus avoiding ISO's ban on the calculation of analytic total error.
Step 11. Monitor failures
In industry, a standard tool for doing this is called failure reporting and corrective action system (FRACAS). FRACAS is particularly valuable in connection with risk management and the commonly used failure modes and effects analysis (FMEA) tool as it provides field data that can be used to determine the actual frequency of occurrence of various failure modes that are targeted in risk-based QC Plans. Use of such tools is limited in medical laboratories, but each laboratory must develop and implement measures to monitor conformance with requirements.
According to ISO 15189, laboratories must identify and control nonconformities. A non-conformity is defined as nonfulfillment of a requirement. In practical terms, that means errors or defective results due to failures in the pre-examination, examination or postexamination processes. Laboratories need to carefully monitor the results from SQC, PT/EQA programmes, customer complaints and other selected quality indicators. Specific quality indicators are now recommended by many professional organizations and CLSI has published a guideline for development and implementation of quality indicators. 59
It should be noted that the first paper to describe the application of Six Sigma concepts and metrics in a medical laboratory was an assessment of defect rates in pre- and postexamination processes. 60 This application made use of the ‘counting methodology’, where the number of errors or defective results were compared to the total number of opportunities in order to calculate defects per million (DPM), which could then be converted to the sigma-scale by use of standard tables. It is common for laboratories to express error rates in terms of percent, but expression on the Sigma-scale provides a better assessment of the rates that are acceptable. For example, a 5% error rate corresponds to 3.15 sigma, a 1% error rate to 3.95 sigma, a 0.5% error rate to 4.15 sigma, a 0.1% error rate to 4.6 sigma, a 0.05% error rate to 4.75 sigma and a 0.01% error rate to 5.2 sigma. Laboratories goals for error rates should be set at less than 0.01% to achieve world class quality. Plebani et al. 61 recently reviewed data from 2009 to 2013 for quality indicators for the preanalytic phase and documented median sigma values between 4 and 5 for misidentification errors, transcription errors, incorrect sample type, incorrect fill level and unsuitable samples. Much attention has been paid to preanalytic and postanalytic error rates in the last 15 years and much improvement has been made. According to Sikaris, 62 postanalytical quality should be the ultimate control on the quality of the total examination process and the usefulness of test results in the context of intended clinical use.
Step 12. Improve quality and the total QC plan
The purpose of the previous two steps – measuring quality and performance, as well as monitoring failures – is to identify the needs for improvement. This completes the Deming PDCA cycle with actions to improve the quality of the examination process (go to step 1 in Figure 2) or to improve the quality of the total QC plan (go to step 5).
Concluding remarks
We have focused on the technical requirements for implementing a scientifically based quality control process and have followed Deming's guidance for scientific management by use of the PDCA cycle. Healthcare organizations began adopting Deming's principles when implementing total quality management (TQM) in the 1990s. Six Sigma quality management is an evolution of TQM, providing quantitative performance goals in the form of ‘tolerance limits’ and quality goals in the form of Sigma-metrics, or quality on the Sigma-scale. These improvements provide a more quantitative framework and help make quality both more measurable and more manageable.
While our emphasis has been on technical issues and requirements, it is also important to understand Deming's philosophy for successful applications of scientific management. Important points were summarized in the executive summary of Berwick's recent report on ‘Improving the safety of patients in England’:
63
Give the people of the NHS – top to bottom – career-long help to learn, master and apply modern methods for quality control, quality improvement and quality planning. Make sure pride and joy in work, not fear, infuse the NHS.
Quality begins and ends with people. Front-line workers and analysts are critical for interacting with customers and delivering quality services and patient care. Management is responsible for providing the organizational support and structure to help people do the right things right. Management must define quality policies and provide processes, procedures, tools, and training that will ensure delivery of the intended results. The 6σQMS described here will hopefully help you and your laboratory provide clear guidance on how to achieve the quality required for the intended use of the results produced by your examination procedures.
Supplementary material
A glossary of terms, control rules, and abbreviations is available online.
Footnotes
Acknowledgements
This article was prepared at the invitation of the Clinical Sciences Reviews Committee of the Association for Clinical Biochemistry and Laboratory Medicine.
Conflict of interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
Not applicable.
Guarantor
JOW.
Contributorship
JOW and SAW contributed equally to this review.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.