
Abstract
Lipoprotein lipase has long been known to hydrolyse triglycerides from triglycerides-rich lipoproteins. More recently, it has been shown to promote the binding of lipoproteins to various lipoprotein receptors. Evidence is also presented regarding the possible atherogenic role of lipoprotein lipase. In theory, lipoprotein lipase deficiency should help to clarify this question. However, the rarity of this condition means that it has not been possible to conduct epidemiological studies. An alternative approach is to investigate the correlation of lipoprotein lipase with onset of cardiovascular disease in prospective studies in large population-based cohorts. Complementary with this approach, animal models have been used to explore the atherogenicity of lipoprotein lipase expressed by macrophages.
Introduction
Lipoprotein lipase (LPL) (EC 3.1.1.34) is synthesized and secreted in several tissues, such as adipose tissue, skeletal muscle, cardiac muscle and macrophages (MΦ), binding to the vascular endothelial cell surface of the capillary through heparan sulphate. LPL is a member of the lipase gene family, which includes pancreatic lipase, hepatic lipase and endothelial lipase.1–3 The human LPL gene is located on the short arm of chromosome 8 (8p22), is about 35 kb in length, contains 10 exons and encodes an enzyme protein consisting of 448 amino acids.2,4,5 LPL catalyses hydrolysis of triglycerides (TG) from chylomicrons and very low density lipoproteins (VLDL), facilitating the incorporation of free fatty acids into adipocytes,6,7 where they are resynthesised into TG and stored. They are utilized as an energy source in cardiac and skeletal muscle. The NH2-terminal domain of LPL controls its catalytic properties, whereas the COOH-terminal domain modulates substrate specificity.8,9 In the 1990s, a number of researchers established that LPL has an additional bridging function, promoting the binding of different lipoproteins to their receptors.10–14
The dual roles of LPL – promoting lipoprotein metabolism and acting as a ligand for binding of lipoproteins to their receptors – have led to speculation about its possible role in the onset of atherosclerosis. Various approaches have been used to study this role. For example, LPL deficiency provides an obvious means of examining this question. However, this condition is rare (approximately one per million in most populations) and it is not possible to conduct epidemiological studies. Experimental studies have been performed using animal models to explore the atherogenicity of LPL expressed by MΦ. We review here the evidence that LPL plays a role in the onset of atherosclerosis.
Studies which indicate that LPL is proatherogenic
LPL is expressed in atherosclerotic lesions
It was first reported in the 1990s that LPL in atherosclerotic lesions was derived from MΦ;15,16 moreover, differences in MΦ expression of LPL contributed to differences in the development of atherosclerotic plaque formation. 17 Concentrations of LPL protein, activity and mRNA in atherosclerosis-prone mice were found to be much higher (several-fold) than in atherosclerosis-resistant counterparts. Babaev et al. 18 transplanted fetal liver cells (from which MΦ cells are derived) from LPL +/+ mice, LPL −/+ mice and LPL −/− mice into irradiated female mice and examined atherosclerotic lesions of the aorta. Mice transplanted with fetal liver cells from LPL +/+ mice had the most severe atherosclerotic lesions in the aorta, suggesting that MΦ LPL may contribute to the severity of atherosclerotic lesions. Ichikawa et al. 19 compared atherosclerotic lesions in rabbits with over-expressed MΦ-specific human LPL, with lesions in wild-type strains, after giving both groups food containing 0.3% cholesterol. Serum lipids were comparable but atherosclerotic lesions were more prominent in the former than in the latter. In a similar vein, peritoneal MΦ from MΦ-specific LPL −/− mice crossed with apo E −/− mice showed less susceptibility to foam cell formation compared with those from apo E −/− mice, 20 suggesting that MΦ LPL may promote atherosclerosis formation.
Elevated LPL activity and mass in post-heparin plasma in subjects with advanced atherosclerosis
Investigators at the National Institutes of Health examined the volume of total calcific atherosclerosis in the heart and thoracic aorta and coronary artery calcific atherosclerosis in 15 patients with homozygous familial hypercholesterolemia. 21 Both LPL activity and mass correlated with these parameters.
LPL is involved in the formation of atherogenic lipoproteins as an enzyme
LPL hydrolyses TG from chylomicrons and VLDL forming chylomicron remnants and VLDL remnants. These are rich in cholesterol esters 22 and become incorporated into MΦ in vitro. 23 Furthermore, the free fatty acids formed by this LPL action are re-esterified by MΦ. 24 This process promotes cholesterol ester accumulation in MΦ, leading to the transformation of MΦ into foam cells. LPL also converts VLDL into intermediate-density lipoproteins (IDL), converted in turn into low density lipoproteins (LDL) by the action of hepatic lipase. LDL is oxidized in vascular subendothelium, incorporated into MΦ via the scavenger receptor, again contributing to formation of foam cells. 25
LPL function as a ligand, independent of its enzyme activity
In 1991, Beisiegel et al. 10 reported that human and bovine LPL promote binding of chylomicrons to a lipoprotein receptor-related protein (LRP) on the surface of HepG2 cells, independently of its enzyme activity. LPL was subsequently shown to be involved in binding a variety of lipoproteins to individual receptors.11,12 The interaction of LDL with LPL is found to be enhanced by its oxidization.13,14 By these mechanisms LPL promotes lipoprotein accumulation in the arterial subendothelial matrix leading to atherosclerosis. LPL not only promotes binding of lipoproteins to receptors but also promotes adhesion of monocytes to the vascular endothelium.26,27
Studies which indicate that LPL is antiatherogenic
LPL deficiency and atherosclerosis
In LPL deficiency, the elevated lipoproteins (chylomicrons) are too large to penetrate into the vascular endothelium, and plasma LDL cholesterol concentrations are reduced; as a result, development of atherosclerosis is attenuated. 7 Consistent with this, Ebara et al. 28 reported a 66-year-old female with LPL deficiency without any obvious atherosclerotic disease in carotid, femoral and coronary arteries as assessed by ultrasonography and electrocardiography after exercise-tolerance testing. They also reported a 60-year-old female with LPL deficiency (caused by a different mutation), who had no atherosclerotic lesions in her coronary arteries as examined by multidetector computer tomography (MDCT). 29 We have similarly reported a 53-year-old man with LPL deficiency without atherosclerosis. 30
On the other hand, Benlian et al. 31 investigated four patients with profound functional deficiency of LPL associated with reduced enzymatic mass due to missense mutations on both alleles of the LPL gene; in all four, premature atherosclerosis (before age 55) was observed. Similarly, Saiki et al. 32 reported a 55-year-old man with LPL deficiency associated with coronary artery disease and severe systemic atherosclerosis. Furthermore, in heterozygous LPL deficiency, reduced LPL activity is associated with premature atherosclerosis33,34 or the onset of familial combined hyperlipidemia. 35 Finally, Hu et al. 36 reported in a meta-analysis that LPL Asn291Ser mutations were associated with high TG, low HDL and high rates of coronary artery disease. Conversely, LPL mutations leading to increased LPL activity were protective against the development of coronary artery disease in another meta-analysis. 37
Literature referring to lipoprotein lipase deficiency in atherosclerosis.

LPL: lipoprotein lipase.
Animal models of LPL deficiency have been used to examine this issue. Zhang et al. 39 studied LPL-deficient mice, where human LPL gene was introduced at birth with adenoviral vectors. The mice exhibited low HDL-C and marked hypertriglyceridaemia on a normal chow diet. Although at four months of age there were no atherosclerotic lesions of the aorta in the LPL-deficient mice but at 15 months of age more advanced atherosclerotic lesions were observed compared with those in wild-type or with heterozygous LPL deficiency. These findings suggest that as individuals with LPL deficiency age, atherosclerotic lesions may progress. The authors of this study proposed that chylomicron derived from LPL-deficient subjects may contribute to increasing the expression of VCAM-1 and MCP-1 from endothelial cells.
Atherosclerosis in animal models over-expressing LPL
In animal models, over-expressing human LPL is associated with improved serum lipid profile.40–42 Shimada et al. 41 found that over-expression of human LPL in LDL receptor −/− mice was associated with a significant reduction of aortic atherosclerotic lesions, as well as decreases in TG and remnants in plasma, compared with LDL receptor−/− mice without human LPL over-expression. Tsutsumi et al. 43 found that NO-1886, a compound that increases LPL activity, was associated with elevated HDL-C and reduced TG, and that its long-term (90-day) administration inhibits atherogenesis in the coronary arteries of rats with experimental atherosclerosis. Fan et al. 42 found that when transgenic rabbits expressing human LPL were fed a cholesterol-rich diet, the development of hypercholesterolaemia and aortic atherosclerosis was dramatically suppressed. Thus, systemically increased LPL activity affects the metabolism of all classes of lipoproteins, and plays a crucial role in plasma TG hydrolysis and lipoprotein conversion. Over-expression of LPL appears to protect against diet-induced hypercholesterolaemia and atherosclerosis.
Studies suggesting serum LPL protein concentration is a useful biomarker predicting cardiovascular disease
In clinical practice, LPL used to be quantified by measuring its activity in post-heparin plasma (PHP) using isotope-labelled substrate. In 1993, we established a sandwich enzyme-linked immunoassay (EIA) system for quantifying LPL protein concentration in PHP, using antibovine milk LPL monoclonal antibody and antibovine milk LPL polyclonal antibody (Figure 1(a)).
44
Subsequently, the clinical significance of measuring LPL concentrations in serum rather than PHP was clarified.45,46 Hanyu et al.
47
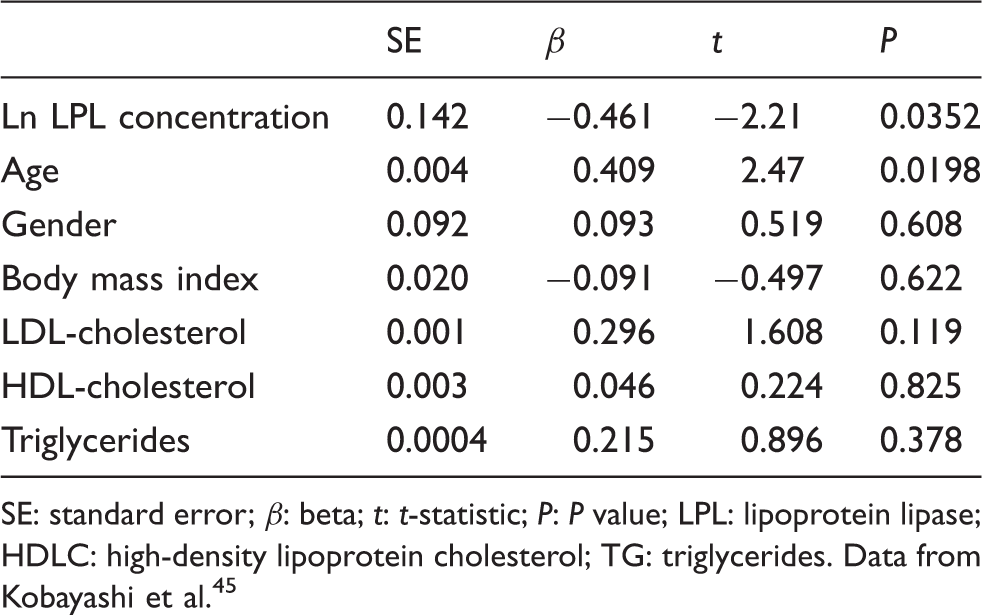
found that serum LPL mass correlated significantly with insulin sensitivity analysed by minimal modelling, regardless of whether the subjects had normal glucose tolerance, impaired glucose tolerance or diabetes. Hitsumoto et al.48,49 found that men with coronary atherosclerosis had significantly lower pre-heparin LPL mass than did men without atherosclerosis or healthy men. Thus, LPL mass appears to be an independent determinant of coronary artery disease48,49 even after adjusting for metabolic parameters, including serum TG and HDL-C. We examined the correlation of intima-media thickness (IMT) of the carotid artery by ultrasonography and serum LPL concentration in dyslipidemic patients. There was an inverse correlation between serum LPL concentration and IMT, independent of age, gender, body mass index, LDL-C, HDL-C and TG (Table 2). In the EPIC-Norfolk population cohort, reduced serum LPL activity was associated with an increased risk of coronary artery disease.
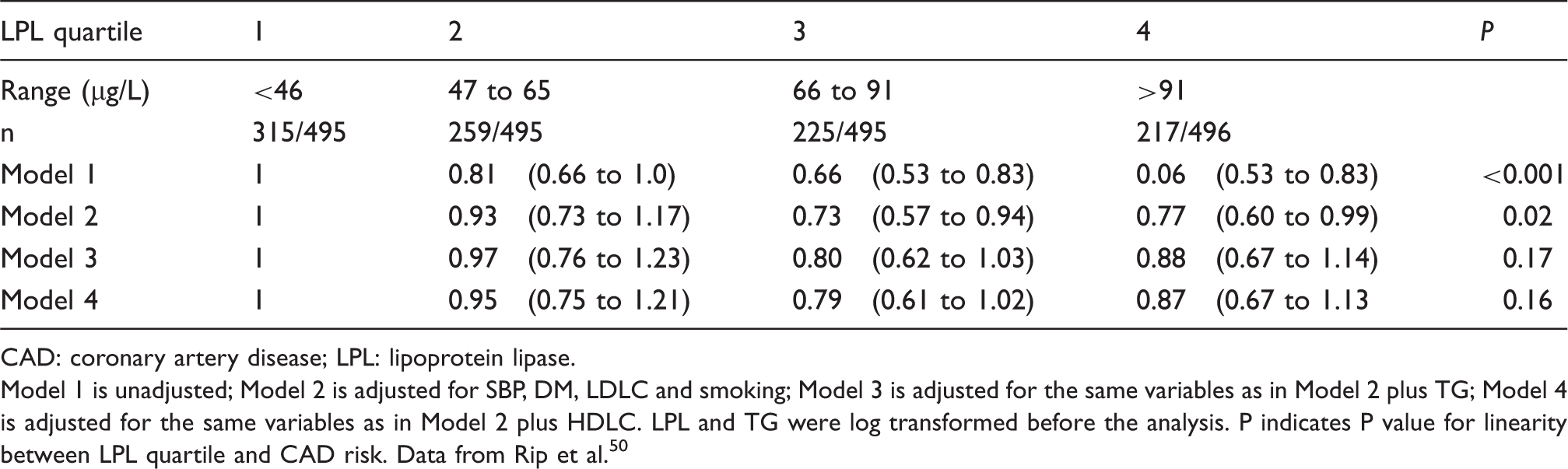
50
This effect remained significant after adjustment for blood pressure, diabetes, smoking, body mass index and LDL-C but not after additional adjustment for high-density HDL-C or TG.
50
(a) ELISA for quantifying human LPL protein concentration in either serum or PHP using antibovine milk LPL monoclonal antibody and antibovine milk LPL polyclonal antibody.
44
(b) ELISA for quantifying human LPL protein concentration using two different monoclonal antibodies against human LPL (57A5 and 88B8) for the sandwich ELISA.
45
A multiple regression analysis on the relationship of intima media thickness of common carotid artery assessed by ultrasonography to LPL, age, gender, body mass index, LDLC, HDLC and TG. SE: standard error; β: beta; t: t-statistic; P: P value; LPL: lipoprotein lipase; HDLC: high-density lipoprotein cholesterol; TG: triglycerides. Data from Kobayashi et al.
45
Odds ratios for future CAD according to serum LPL concentration quartile. CAD: coronary artery disease; LPL: lipoprotein lipase. Model 1 is unadjusted; Model 2 is adjusted for SBP, DM, LDLC and smoking; Model 3 is adjusted for the same variables as in Model 2 plus TG; Model 4 is adjusted for the same variables as in Model 2 plus HDLC. LPL and TG were log transformed before the analysis. P indicates P value for linearity between LPL quartile and CAD risk. Data from Rip et al.
50

Recently, Shirakawa et al. 51 developed a new high-sensitivity LPL measurement system using two different antihuman LPL monoclonal antibodies (Figure 1(b)). They found that remnant-like particles associated with cholesterol (RLP-C) or triglycerides (RLP-TG) and RLP-TG/RLP-C ratio correlated inversely with serum LPL protein concentrations. Given that RLP-TG/RLP-C ratio reflects the particle size of the RLP, 52 this raises the possibility that LPL may play an important role in remnant metabolism.
Conclusion
This review has summarized various strands of evidence examining the relationship between LPL and atherosclerosis. Overall, the evidence is inconclusive. Large-scale prospective cohort studies of the relationship between serum LPL concentration and cardiovascular disease in various populations may help to resolve this question.
Footnotes
Acknowledgements
We thank Dr Katsuyuki Nakajima of Gunma University for critically reading the manuscript and Kazuya Miyashita for performing several experiments discussed in this review.
This article was prepared at the invitation of the Clinical Sciences Reviews Committee of the Association for Clinical Biochemistry and Laboratory Medicine.
Declaration of conflicting interests
None.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethical approval
Not applicable.
Guarantor
JK.
Contributorship
Both authors contributed equally to the manuscript.