
Abstract
Background
Familial lipoprotein lipase (LPL) deficiency is a very rare autosomal recessive disorder characterized by marked elevation of plasma triglyceride concentrations. Since 1989, a variety of mutations have been reported in affected patients. Studies on subjects with heterozygous LPL deficiency, on the other hand, have been limited.
Methods
We examined post-heparin plasma LPL activity in 15 subjects with heterozygous LPL deficiency.
Results
The heterozygotes exhibited normal or slightly elevated plasma triglyceride concentrations. The mean LPL activity was reduced by 25% in the heterozygotes relative to controls. Interestingly, LPL activity was reduced specifically in female heterozygotes.
Conclusion
LPL activity is decreased in female, but not in male, subjects heterozygous for a number of different LPL gene mutations.
Keywords
Introduction
Familial lipoprotein lipase (LPL) deficiency is an inherited disorder characterized by marked elevation of plasma triglyceride concentrations due to defective hydrolysis of chylomicron triglycerides. 1 The disorder is extremely rare, occurring in approximately 1 in 1,000,000 persons: heterozygous LPL deficiency is estimated to occur in 1 out of 500 people.
Since the establishment of a selective immunochemical method to measure post-heparin plasma LPL activity, 2 the detection of heterozygous carriers of LPL deficiency has become of great interest. The aim of this study was to search for biochemical markers to detect carriers.
Subjects and methods
Fifteen parents (eight fathers and seven mothers) of eight previously reported patients (probands) with familial LPL deficiency were subjects for the study.3–6
The eight probands had plasma triglyceride (TG) concentrations ranging between 17.25 and 215.86 mmol/L. The post-heparin plasma LPL activity for each patient was less than 1.2 µmol free fatty acids (FFA)/mL/h, 20% of the mean activity in control subjects (Using our quantitative method, mean LPL activity for adult subjects was 6.42, and the lower limit of detectable LPL activity was 0.6 µmol FFA/ml/h. 5 ). Six of the probands were infants, including two males and four females, and the remaining two were adults. The probands had heterogeneous mutations in the LPL gene, including missense mutations in three patients, nonsense mutations in two, donor splice-site mutation in one, and the remaining two identified as compound heterozygotes.
Characteristics of the obligate heterozygous subjects with familial lipoprotein lipase deficiency.
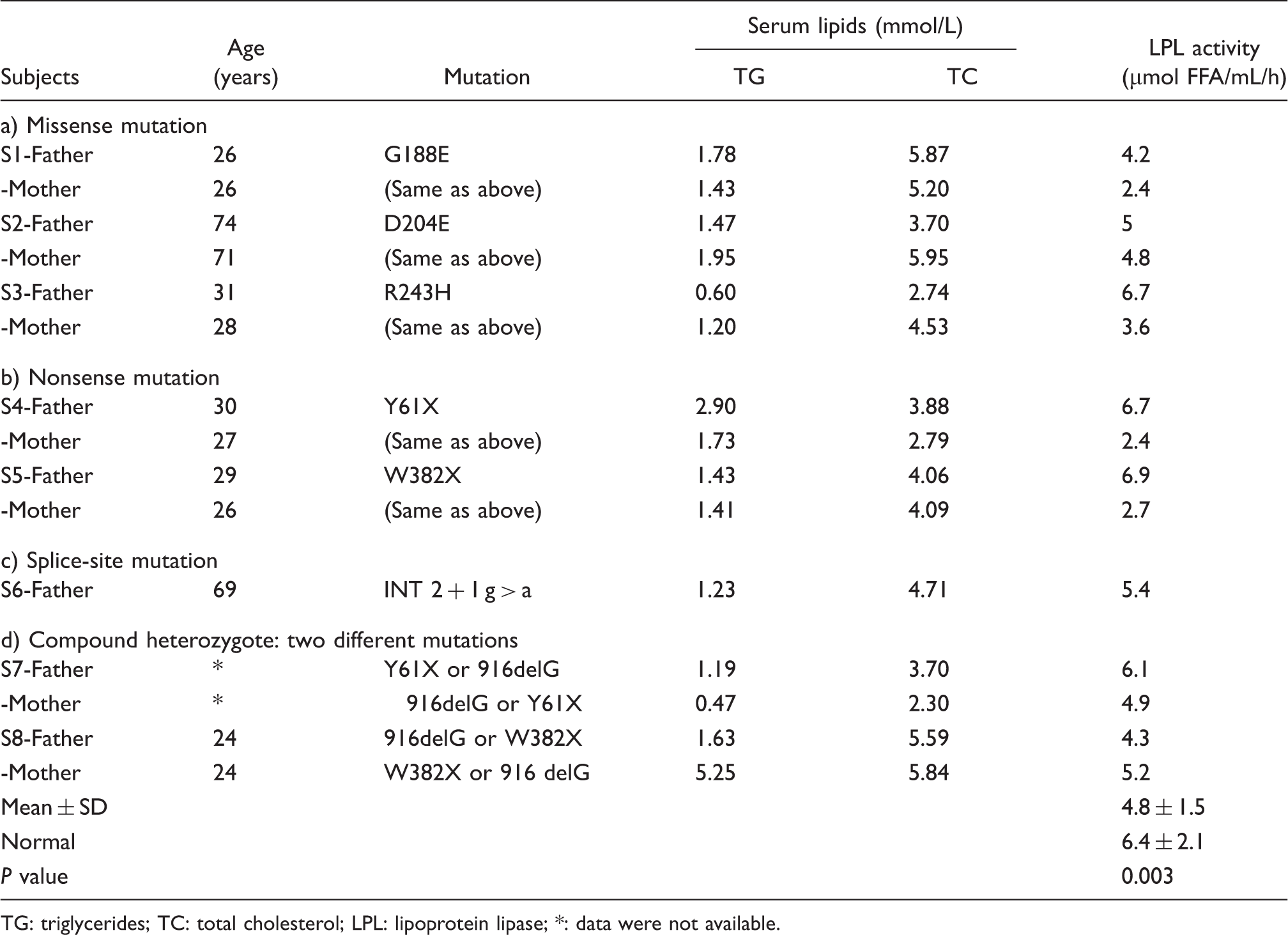
TG: triglycerides; TC: total cholesterol; LPL: lipoprotein lipase; *: data were not available.
Plasma lipids and post-heparin plasma LPL activity were measured after a 14-h fast. The LPL activity was determined selectively by an immunochemical method using antiserum specifically directed against hepatic lipase, as described previously. 2
Results were expressed as means ± standard deviation (SD). Values in the obligate heterozygote parents were compared with those in the reference controls by a two-tailed Student’s t-test. P values < 0.05 were considered statistically significant.
Results
Plasma lipid concentrations
Of the 15 heterozygote parents, 10 had normal plasma TG concentrations (<1.69 mmol/L): only two showed plasma TG concentrations of over 2.26 mmol/L (Table 1). No parent showed elevation of plasma total cholesterol (TC) concentrations (<6.21 mmol/L).
Post-heparin plasma LPL activity
The post-heparin plasma LPL activity in 25 normolipidemic (TC < 6.21 mmol/L and TG < 1.69 mmol/L) control subjects, 13 males and 12 females, averaged 6.4 ± 2.1 µmol FFA/ml/h. 2 There was no significant gender difference in the control group, with an average LPL activity of 6.1 ± 2.1 and 6.8 ± 2.2 in males and females, respectively (P = 0.409). 2
The average LPL activity in the obligate heterozygote parents was significantly lower than that of the control subjects (Table 1); the mean enzyme activity was 75% of that in the control subjects (4.8 ± 1.5 and 6.4 ± 2.1 µmol FFA/mL/h; P = 0.011). Although the mean LPL activity of the heterozygotes was significantly lower than that of the control subjects, there was a great degree of overlap; 10 heterozygotes had normal LPL activity ([mean-SD]), and five heterozygotes had decreased LPL activity. The LPL activity did not differ depending on the type of mutation; the activity levels (µmol FFA/mL/h) were 4.5 ± 1.4 in the six heterozygotes with missense mutations, 4.7 ± 2.5 in the four with nonsense mutations, and 5.4 in the heterozygote with the splice-site mutation.
The average LPL activity was significantly lower in females than in males (3.7 ± 1.2 vs. 5.7 ± 1.1, P = 0.014). We compared the LPL activity of male and female heterozygotes with that of the control subjects of their respective gender. Female heterozygotes showed reduced enzyme activity compared to the female controls (3.7 ± 1.2 vs. 6.8 ± 2.2, P = 0.003), while LPL activity of male heterozygotes was comparable to male control subjects (5.7 ± 1.1 vs. 6.1 ± 2.1, P = 0.609).
Discussion
In search for biochemical markers for heterozygous carriers of LPL deficiency, we collected data on the heterozygote parents of known probands.3–6 In contrast to the marked elevation of the plasma TG concentrations in the probands, the carrier parents exhibited normal or only mild to moderate increases in their plasma TG concentrations. The mean post-heparin plasma LPL activity in the heterozygotes was 25% lower than that in the control subjects. The average LPL activity was decreased in female, but not in male, carriers.
Most previous reports indicate that heterozygotes for LPL deficiency show normal or mild to moderate hypertriglyceridemia. Our observation was consistent with these reports.
Brunzell et al. 1 reported that obligate heterozygote parents and other family members of LPL-deficient patients show normal or reduced post-heparin plasma LPL activity. However, non-heterozygous subjects may also have been included as study subjects in that report. Since the first mutation in the LPL gene was reported, 7 several conflicting results have been published with a limited number of subjects. 1 Our observation of a 25% reduction of post-heparin plasma LPL activity in 15 carriers with different types of LPL gene mutations was mostly consistent with previously published reports. 1 Although the reduction was statistically significant, we found overlap between the carriers and reference control subjects. Thus, plasma LPL activity does not fulfill the necessary criteria for a reliable biochemical marker to identify subjects with heterozygous LPL deficiency.
Interestingly, the average LPL activity was decreased only in the female carriers. In our study, six of the eight probands were infants under one year of age, and the female carriers were breastfeeding the probands at the time of sample collection. Previous reports have demonstrated that LPL activity in the mammary glands is markedly elevated during lactation, and simultaneously reduced in adipose tissues, to enable dietary and stored lipids to be preferentially incorporated into milk fat. 8 The contribution of the mammary gland LPL to the total post-heparin plasma LPL activity remains to be determined. To our knowledge, no reports have addressed the significance of lactating mammary gland LPL in carrier mothers; however, the crucial role of mammary gland LPL was recognized in a lactating female patient with familial LPL deficiency. 9 This observation raises an intriguing question for future research, which should be performed on a larger subject population.
To date, no biochemical markers have been identified that can detect carriers of familial LPL deficiency in the general population. During our study, we made an intriguing observation that LPL activity is decreased specifically in female subjects heterozygous for LPL gene mutations. This gender difference remains to be further examined.
Footnotes
Declaration of conflicting interests
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Funding
None declared.
Ethical approval
Ethical Committee of Toranomon Hospital approved this study (No. 658).
Guarantor
TM.
Contributorship
TM contributed to the measurement of enzyme activities of all study subjects and conceived this work. TE and MO contributed to the preparation of the manuscript.