
Abstract
Hypertriglyceridaemia is a common biochemical abnormality that can be due to primary causes or, more commonly, secondary causes. Moderate hypertriglyceridaemia is a risk factor for cardiovascular disease and can develop into severe hypertriglyceridaemia which is a risk factor for acute pancreatitis. Familial chylomicronaemia is a rare autosomal recessive disorder, usually diagnosed in childhood and is characterized by marked hypertriglyceridaemia and biochemical deficiency of lipoprotein lipase (LPL), apolipoprotein (apo) C-II, homozygous (or compound heterozygous) gene mutations in LPL or more rarely, APOC2. Recently, loss-of-function mutations in the APOA5 gene have been reported in patients with severe hypertriglyceridaemia in whom LPL or APOC2 mutations were not found. We describe the clinical features and genetic analysis of three patients with severe hypertriglyceridaemia including novel mutations LPL c.464T>C (p.Leu155Pro) and APOA5 c.823C>T (p.Gln275*).
Introduction
Hypertriglyceridaemia (fasting triglyceride greater than the 95th percentile for age and sex) is a common biochemical abnormality that can be due to primary causes or, more commonly, secondary causes. 1 Hypertriglyceridaemia is likely to be a cardiovascular risk factor independent of high-density lipoprotein (HDL) cholesterol, 2 with severe hypertriglyceridaemia (fasting triglyceride >10 mmol/L) increasing the risk of acute pancreatitis. 3 Consistent with these findings, a recent meta-analysis found an association between fasting hypertriglyceridaemia and risk of cardiovascular death, myocardial infarction and possibly acute pancreatitis. 4
Lipoprotein lipase (LPL; EC 3.1.1.34) is the rate-determining enzyme in the hydrolysis of the triglyceride core in circulating postprandial triglyceride-rich lipoprotein (TRL) particles. 5 Catalytically active LPL is a non-covalent homodimeric glycoprotein anchored to heparin sulphate proteoglycans on the luminal plasma membrane of capillary endothelium, where it hydrolyses TRL and delivers free fatty acids to parenchymal cells. LPL has an absolute requirement for its cofactor, apolipoprotein (apo) C-II.
Familial chylomicronaemia is a rare autosomal recessive disorder usually diagnosed in childhood, characterized by marked hypertriglyceridaemia and biochemical deficiency of LPL, apoC-II, homozygous (or compound heterozygous) gene mutations in LPL or more rarely, APOC2.6,7 ApoA-V is a potent modulator of triglyceride metabolism via an LPL stimulatory mechanism which is present at very low levels in the circulation. 8 Inheritance of rare loss-of-function mutations in the APOA5, LMF1 and GPIHBP1 genes can also cause severe hypertriglyceridaemia. 9 Clinical manifestations of familial chylomicronaemia can include recurrent episodes of epigastric pain, pancreatitis, eruptive cutaneous xanthomata, hepatosplenomegaly and lipaemia retinalis. There does not appear to be increased risk of cardiovascular disease, although premature atherosclerosis has been reported in patients with LPL deficiency. 10
We describe the clinical features and genetic analysis of three patients with severe hypertriglyceridaemia, including novel mutations LPL c.464T>C (p.Leu155Pro) and APOA5 c.823C>T (p.Gln275*).
Case reports
Case 1
A 42-year-old woman was referred to a lipid clinic by her general practitioner for management of hyperlipidaemia. The patient had a history of recurrent admissions to hospital for acute pancreatitis associated with marked hypertriglyceridaemia. There was no history of diabetes mellitus, renal or hepatic dysfunction, systemic disease or excess alcohol consumption.
Elevated triglyceride levels were first observed during investigation of neonatal jaundice. Her first admission for acute pancreatitis was at the age of 16 years, during mumps infection, followed by five episodes up to the date of presentation, with documented triglyceride and lipase concentrations up to 139 mmol/L and 1110 U/L, respectively. During these admissions, standard treatment with nil orally, intravenous fluids and pain relief was prescribed. Abdominal ultrasonography showed a bulky, homogeneous pancreas, but no evidence of gallstones.
She had experienced one episode of transient blurred vision associated with weakness and numbness of the left arm in 2008, after which her oral contraceptive was ceased and low-dose aspirin commenced. Other medications at that time comprised atorvastatin, which was discontinued because of myalgia, and n-3 fatty acids with which compliance was poor. She had previously been treated with gemfibrozil, but this was ceased because of liver dysfunction.
She was separated from her husband and lived alone with one son aged 12 years, whose lipid profile was normal. A brother and sister were also normolipidaemic. Her mother died at the age of 38 years from hepatitis B infection, and her father at the age of 55 years from motor neurone disease. A maternal grandmother died aged 64 years of myocardial infarction. However, the lipid profiles of these relatives were unknown.
On examination, she was 51 kg in weight and 1.52 m in height, with a body mass index (BMI) of 22.1 kg/m2. She appeared well, with no peripheral stigmata of lipid disorders or signs of endocrine or liver disease. A waist circumference was 77 cm, blood pressure 140/75 mmHg and her clinical examination unremarkable.
Plasma lipids on presentation, clinical findings and genetic analysis.
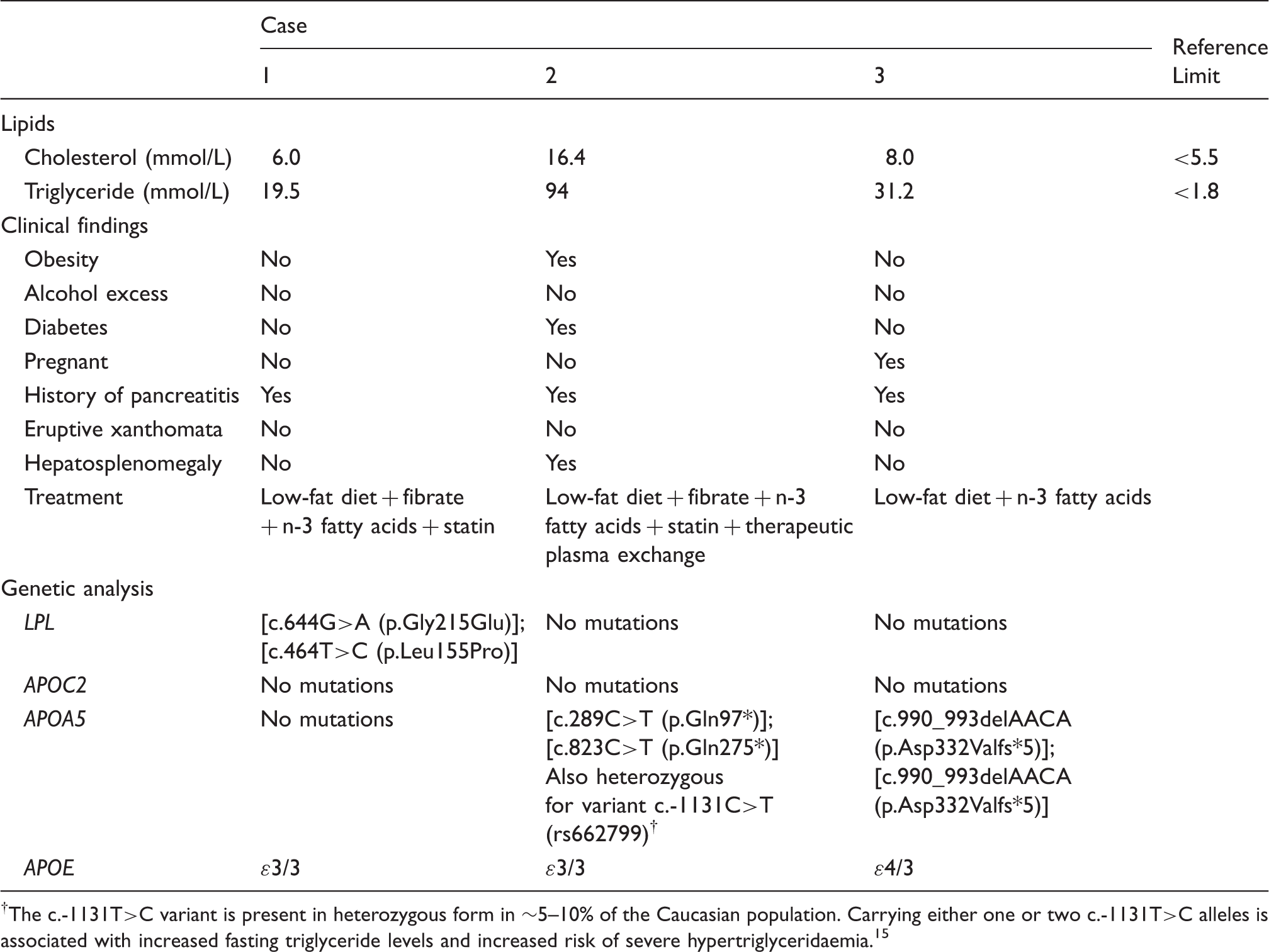
The c.-1131T>C variant is present in heterozygous form in ∼5–10% of the Caucasian population. Carrying either one or two c.-1131T>C alleles is associated with increased fasting triglyceride levels and increased risk of severe hypertriglyceridaemia. 15
Exon-by-exon sequence and splice site analysis of the LPL, APOC2 and APOA5 genes, and the region of the apoA5 promoter containing -1131T>C, revealed that the patient was compound heterozygous for c.644G>A (p.Gly215Glu) and a novel mutation c.464T>C (p.Leu155Pro) in the LPL gene.
Case 2
A 70-year-old Caucasian woman was referred to a renal clinic for assessment of hypertension. The patient had a history of hypertriglyceridaemia, type 2 diabetes with axonal neuropathy and uncontrolled hypertension. A lipid profile showed a total cholesterol and triglyceride of 16.4 mmol/L and 94 mmol/L, respectively (Table 1). She reported two previous episodes of acute pancreatitis and was noted to have milky plasma during an admission for the birth of twins.
Triglyceride concentrations of exceeding 20 mmol/L had persisted over many years, despite diet modification and lipid-lowering therapies. She denied alcohol intake and was non-smoker. Her medications included candesartan, moxonidine, rosuvastatin, fenofibrate, spironolactone and n-3 fatty acids. Her diabetes was managed with metformin and insulin with HbA1c levels ranging from 46 to 64 mmol/mol. There was no history of cardiovascular or cerebrovascular insufficiency and a coronary angiogram performed in 2008 was normal. Her father died prematurely at the age of 47 years from heart disease, and her mother died at the age of 80 years from bladder cancer. She had six children and there was no known family history of lipid disorders.
On examination, she was 81 kg in weight and 1.56 m in height, with a BMI of 33.3 kg/m2. She appeared well, with no peripheral stigmata of lipid disorders or signs of endocrine or liver disease. Her blood pressure was 130/80 mmHg, she was in sinus rhythm at 72 beats/min and her heart size and sounds were normal with no significant murmurs. There was no evidence of heart failure. Peripheral pulses were normal, there were no carotid or abdominal bruits and her chest was clear. Abdominal examination showed hepatosplenomegaly, with no masses, ascites or abdominal tenderness.
A computed tomography angiogram showed right renal artery stenosis for which she underwent angioplasty and stenting, a 3.4 cm abdominal aortic aneurysm, splenomegaly and portal hypertension. Liver function tests and lipase were normal. Given her severe hypertriglyceridaemia and high risk for pancreatitis, she commenced therapeutic plasma exchange on a weekly basis. In addition, she received fenofibrate (145 mg daily), rosuvastatin (10 mg/day) and n-3 fatty acids (6 g daily in divided doses).
Sequence analysis of the LPL, APOC2 and APOA5 genes identified two nonsense mutations in APOA5, c.289C>T (p.Gln97*) and the novel c.823C>T (p.Gln275*).
Case 3
A 35-year-old Burmese woman was incidentally noted to have milky peritoneal fluid at Caesarean section. Biochemical testing showed a total cholesterol and triglyceride of 8.0 mmol/L and 31.2 mmol/L, respectively, with a normal lipase at 39 U/L. At 3-month post-partum review, fasting triglycerides were 7.1 mmol/L, she was commenced on low-dose n-3 fatty acids (3 g daily) and referred to a lipid disorders clinic.
On clinic review at six months post-partum, she could not recall the presence of any eruptive skin lesions, but did report several episodes of epigastric pain over the past years, the first occurring in her teens. At age 23, she was hospitalized for two weeks in Burma with severe epigastric pain radiating through to her back that was associated with vomiting and which required intravenous fluids, although she was unable to recall the specific diagnosis. She was a non-smoker, did not drink alcohol, was breast feeding and had discontinued the n-3 fatty acids pending clinic review.
On examination, she was 47 kg in weight and 1.45 m in height, with a BMI of 22 kg/m2. She appeared well, with no peripheral stigmata of lipid disorders or signs of endocrine or liver disease. Her blood pressure was 120/70 mmHg, she was in sinus rhythm at 72 beats/min and her heart size and sounds were normal with no significant murmurs. There was no evidence of heart failure. Peripheral pulses were normal, and there were no carotid or abdominal bruits. Her chest was clear to auscultation, and there were no abdominal organomegaly, masses, ascites or tenderness.
Biochemical testing showed total cholesterol and triglycerides of 6.8 mmol/L and 12.6 mmol/L, respectively, with normal liver function and lipase level (Table 1). She was recommenced on n-3 fatty acids (6 g daily in divided doses) with good effect; a subsequent fasting lipid profile showed total cholesterol and triglycerides of 4.7 mmol/L and 1.8 mmol/L, respectively.
Sequence analysis of the LPL, APOC2 and APOA5 genes revealed that the patient was homozygous for APOA5 c.990_993delAACA (p.Asp332Valfs*5).
Discussion
Hypertriglyceridaemia is most commonly due to secondary causes including obesity, metabolic syndrome, alcohol excess, diabetes mellitus, pregnancy and a variety of drugs. 1 Moderate hypertriglyceridaemia, a risk factor for cardiovascular disease, can develop into severe hypertriglyceridaemia, a risk factor for acute pancreatitis. In severe hypertriglyceridaemia, chylomicrons and very low density lipoprotein are typically increased as both compete for the same clearance mechanism, namely LPL. However, a molecular basis for primary hypertriglyceridaemia is only found in a minority of cases (typically <5%).
Case 1 had long-standing hypertriglyceridaemia first noted as a neonate with a history of recurrent episodes of pancreatitis in the absence of secondary causes and despite a low-fat diet and multiple lipid-lowering agents. She was found to be compound heterozygous for c.644G>A (p.Gly215Glu) and a novel mutation c.464T>C (p.Leu155Pro) in the LPL gene. The LPL Leu155Pro mutation (Leu128 in the mature protein) is predicted to be pathogenic, using in silico algorithms Mutation Taster and PolyPhen2. The LPL Gly215Glu mutation (Gly188Glu in the mature protein) has been well characterized in the literature and has been demonstrated to be catalytically inactive in vitro. 11
Case 2 was obese, had well-controlled type 2 diabetes, difficult to control hypertension due to right renal artery stenosis and long-standing hypertriglyceridaemia first noted peripartum with subsequent episodes of pancreatitis. We identified two nonsense mutations in APOA5, c.289C>T (p.Gln97*) and c.823C>T (p.Gln275*). The apoA-V Gln97* mutation has previously been reported in homozygous form in a 17-year-old male patient with a high triglyceride and low-HDL cholesterol concentration, who had suffered from severe hypertriglyceridaemia and eruptive xanthomas since the age of 2 years. 12 ApoA-V was not detected in the patient’s plasma, suggesting that Gln97* is a null mutation causing complete apoA-V deficiency. The apoA-V Gln275* nonsense mutation has not been previously reported, but is predicted to result in a truncated apoA-V missing ∼25% of the C-terminus. While apoA-V Gln275*, if synthesized, would contain the LPL activation (residues 192–238) and heparin binding (residues 186–227) domains, it is predicted to show decreased lipid binding. In vitro studies showed that an apoA-V deletion mutant of amino acids 301 to 343 exhibited significantly reduced lipid binding activity. 13
Case 3 was detected peripartum at the time of Caesarean section, and although not confirmed, she also likely has had several recurrent episodes of pancreatitis. Apart from pregnancy, there were no secondary causes of hypertriglyceridaemia present. She was shown to be homozygous for APOA5 c.990_993delAACA (p.Asp332Valfs*5). The apoA-V frameshift mutation p.Asp332Valfs*5 is predicted to result in a truncated protein missing ∼10% of the C-terminus. Recent in vitro studies suggest that this apoA-V variant has normal LPL activation, but impaired interactions with heparin, sortilin and the receptor SorLA/LR11. 14 Our study is the first report of a homozygote for APOA5 Asp332Valfs*5 and confirms the pathogenicity of this mutation. Of interest, in the only other report of this mutation, it occurs in compound heterozygous form together with apoA-V Gln97*. 14 APOA5 mutations may be more common than anticipated. Two common APOA5 variants occur in heterozygous form in ∼5–10% of the Caucasian population, c.-1131T>C (rs662799) and p.S19W (rs3135506), each; and carrying one or two of these alleles is associated with increased fasting triglyceride levels and increased risk of severe hypertriglyceridaemia. 15
The APOE genotype influences plasma triglyceride concentrations, with the ɛ2 allele being associated with higher levels than the more common ɛ3 allele. 16 However, none of our cases harboured the ɛ2 allele.
The non-pharmacological treatment of hypertriglyceridaemia includes weight loss, diet modification and exercise, with reduction or discontinuation of alcohol intake. Fibrates remain the cornerstone of pharmacological treatment of hypertriglyceridaemia and the restriction of dietary fat and/or use of fibrates can prevent the progression to acute pancreatitis. As shown in cases 1 and 2, combination pharmacotherapy, e.g. fibrates, niacin, n-3 fatty acids alone or in combination with statins, with appropriate biochemical monitoring may be required for refractory severe hypertriglyceridaemia. Severe hypertriglyceridaemia during pregnancy, especially when complicated with pancreatitis, carries a significant risk of mortality for both the mother and the foetus. 17
In conclusion, we describe the clinical features and genetic analysis of three patients with severe hypertriglyceridaemia including novel mutations LPL c.464T>C (p.Leu155Pro) and APOA5 c.823C>T (p.Gln275*).
Footnotes
Acknowledgements
We are grateful to Ms Lan Nguyen for her assistance with DNA sequence analysis.
Declaration of conflicting interests
None.
Funding
JRB is supported by a Practitioner Fellowship from the Royal Perth Hospital Medical Research Foundation.
Ethical approval
Written informed consent from the patients was obtained.
Guarantor
JRB.
Contributorship
IH-C and JRB conceived the study. JK, IH-C and JRB recruited patients and analysed data. AJH analysed the genetic data and wrote the first draft of the manuscript. All authors reviewed and edited the manuscript and approved the final version of the manuscript.