
Abstract
Acute kidney injury is common, dangerous and costly, affecting around one in five patients emergency admissions to hospital. Although survival decreases as disease worsens, it is now apparent that even modest degrees of dysfunction are not only associated with higher mortality but are an independent risk factor for death. This review focuses on the pathophysiology of acute kidney injury secondary to ischaemia – its commonest aetiology. The haemodynamic disturbances, endothelial injury, epithelial cell injury and immunological mechanisms underpinning its initiation and extension will be discussed along with the considerable and complex interplay between these factors that lead to an intense, pro-inflammatory state. Mechanisms of tubular recovery will be discussed but also the pathophysiology of abnormal repair with its direct consequences for long-term renal function. Finally, the concept of ‘organ cross-talk’ will be introduced as a potential explanation for the higher mortality observed with acute kidney injury that might be deemed modest in conventional biochemical terms.
Keywords
Introduction
Acute kidney injury (AKI) is common, affecting around one in five patients emergency admissions to hospital 1 and dangerous, with contemporary series reporting mortality rates of around 25–40% with the most severe disease.2,3 Although survival decreases, progressively, as disease worsens, it is now apparent that even very modest acute elevations in serum creatinine (e.g. 26.5 µmol/L within 48 h) are associated with higher mortality. 4 Evidence also suggests that patients are not simply dying with AKI as a fellow traveller but because of it. 5 Rather than uraemia or its attributable complications (e.g. hyperkalaemia), common causes of death include bleeding and sepsis. 6
Survivors often fail to recover renal function 7 with significant numbers needing long-term dialysis.8,9 Long-term survival may be reduced especially in those with persisting renal dysfunction.8,9 AKI also carries a significant economic impact; severe disease often requires expensive interventions such as dialysis or intensive care, but even modest AKI (with a serum creatinine rise ≥ 44 µmol/L) has been shown to increase hospital length of stay by 3.5 days and hospital costs by approximately £4800. 10
Common, dangerous and costly, it is therefore unfortunate that the recognition and subsequent management of AKI and patients at risk of it are often poor.11,12 It is frequently the very basics of care that are neglected; attention to patients’ volume status, medication review, and recognition and treatment of the underlying cause of AKI, for instance 13 reflecting not just clinical and organizational deficiencies but also a failure to appreciate the pathophysiology of those at risk of AKI and those developing it. There appears to be a clear knowledge gap in practitioners’ understanding of the condition as a whole 14 that requires urgent attention. Quite how these clinical, organizational and educational deficiencies should be addressed remains unclear though. 15
K-DIGO classification for the diagnosis and staging of acute kidney injury. 16 AKI is defined as any of the following: • Increase in SCr by ≥26.5 µmol/L (≥0.3 mg/dL) within 48 h; or • Increase in SCr to ≥1.5 times baseline, which is known or presumed to have occurred within the prior seven days or • Urine volume < 0.5 mL/kg/h for 6 h where SCr = serum creatinine. The severity of AKI is staged as follows, using the worst* SCr or urine output stage over a given period of time (usually seven days).

where eGFR =estimated GFR.
For example, a doubling of SCr from baseline with a normal urine output would be staged as AKI 2.
Acute renal dysfunction may be divided into three main categories: pre-renal azotaemia, intrinsic AKI and post-renal (obstructive) AKI. Intrinsic AKI may be further sub-divided into glomerular causes (e.g. rapidly progressive glomerulonephritis), vascular causes (e.g. scleroderma renal crisis) and tubulo-interstitial disease. Most tubulo-interstitial disease and indeed most AKI are caused by ischaemia. 19 It is the pathophysiology of this entity, ischaemic AKI, which contributes so much to morbidity, mortality and cost, which will be examined in this review. The review will also draw on clinical correlates, particularly in relation to mortality and long-term renal outcomes.
Ischaemic AKI can be represented as one end of a pathophysiological spectrum separated from purely pre-renal disease (an appropriate response to renal hypoperfusion, with intense sodium and water retention) by an increasing degree of renal cell injury. As will be shown, later, there is no set threshold at which renal cell injury occurs, with different parts of the kidney carrying differing susceptibilities to hypoperfusion. Nevertheless, the initiating insult may be so profound that the pre-renal phase is short-lived and, effectively, irreversible. Conversely, in many cases, timely intervention to restore renal perfusion may mitigate the severity of evolving ischaemic AKI by preventing still functioning tissue (i.e. parts of the kidney still in the pre-renal phase) from progressing to overt injury.
Although a localized or generalized reduction in blood flow is the common pathway into the pathophysiological disturbances of ischaemic AKI, these can arise from several different sources (Table 2) including the various causes of decreased intravascular volume and hypotension, drug-induced ischaemia and sepsis. Figure 1 gives a schematic representation of the phases of ischaemic AKI as described by Molitoris.
20
Following the initiating insult that leads to the first rapid drop in the glomerular filtration rate (GFR), there follows an extension phase, during which the renal insult is propagated and function declines further. This phase is characterized by an intense ischaemia-reperfusion injury. After this has subsided, renal function remains at a nadir in the ‘maintenance’ phase for a variable length of time before renal recovery occurs. The depth of the decline in renal function, the duration of the maintenance phase and the degree of renal recovery are all factors that vary in clinical practice although the time course of ischaemic AKI, from insult to the first evidence of recovery, is usually in the order of 7 to 21 days.
Schematic representation of the different phases of ischaemic acute kidney injury. Causes of ischaemic acute kidney injury.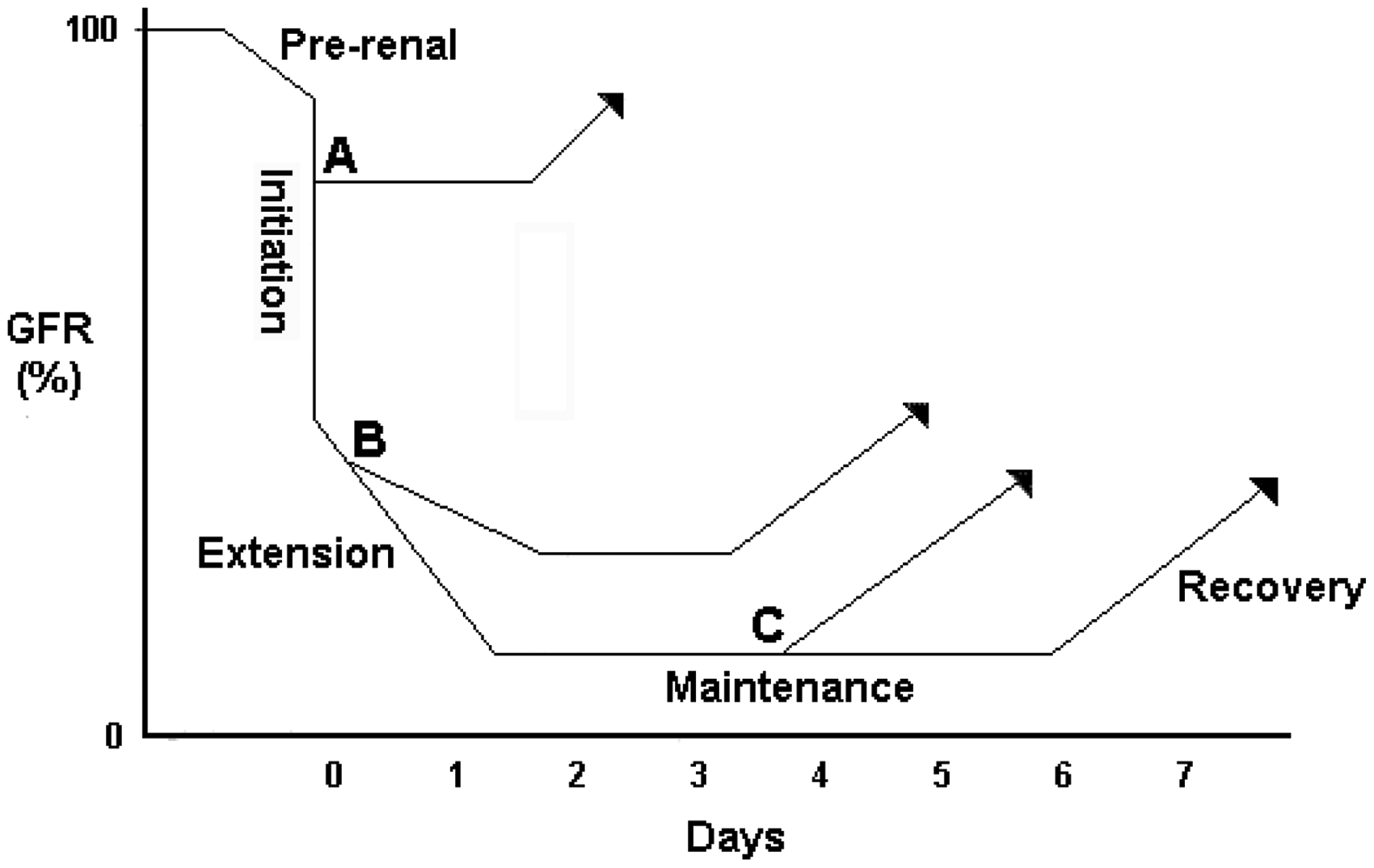

The pathophysiological factors underpinning the initiation and extension phases may be examined under four headings: haemodynamic factors, endothelial injury, epithelial cell injury and immunological factors. Although it is useful to categorize the response in this way, it must be stressed that there is considerable and complex interplay between these different mechanisms, driving and potentiating renal injury.
Haemodynamic factors
Rather than alterations in whole organ perfusion, it may be regional changes in intra-renal blood flow that explain the depth of loss of kidney function in individual instances.
21
The outer medulla, for instance, has been shown to be particularly vulnerable in experimental models22,23 and may be explained by factors including local oedema
24
and its complex capillary network.
25
It is not difficult to envisage how overall changes in glomerular blood flow and specific changes in efferent arteriolar resistance may have a profound impact on the vulnerable, downstream micro-circulation. Compounding this, the outer medulla, even in health, exists on the verge of hypoxia (Figure 2) as its blood flow, arising from the vasa recta, is relatively oxygen deplete after supplying the energy-sapping demands of the counter-current exchange system along the loop of Henle.
26
Furthermore, experimental micro-vascular work has highlighted the potential vulnerability of individual nephrons – and in particular, those in the inner cortex – due to their anatomical dependence on the efferent blood flow from multiple glomeruli.
25
Finally, the proximal tubule may have a further, innate vulnerability with experimental work suggesting that it cannot switch to anaerobic glycolytic metabolism when exposed to antimycin A, an inhibitor of oxidative metabolism.
27
Illustration of blood flow and partial pressure of oxygen within the kidney.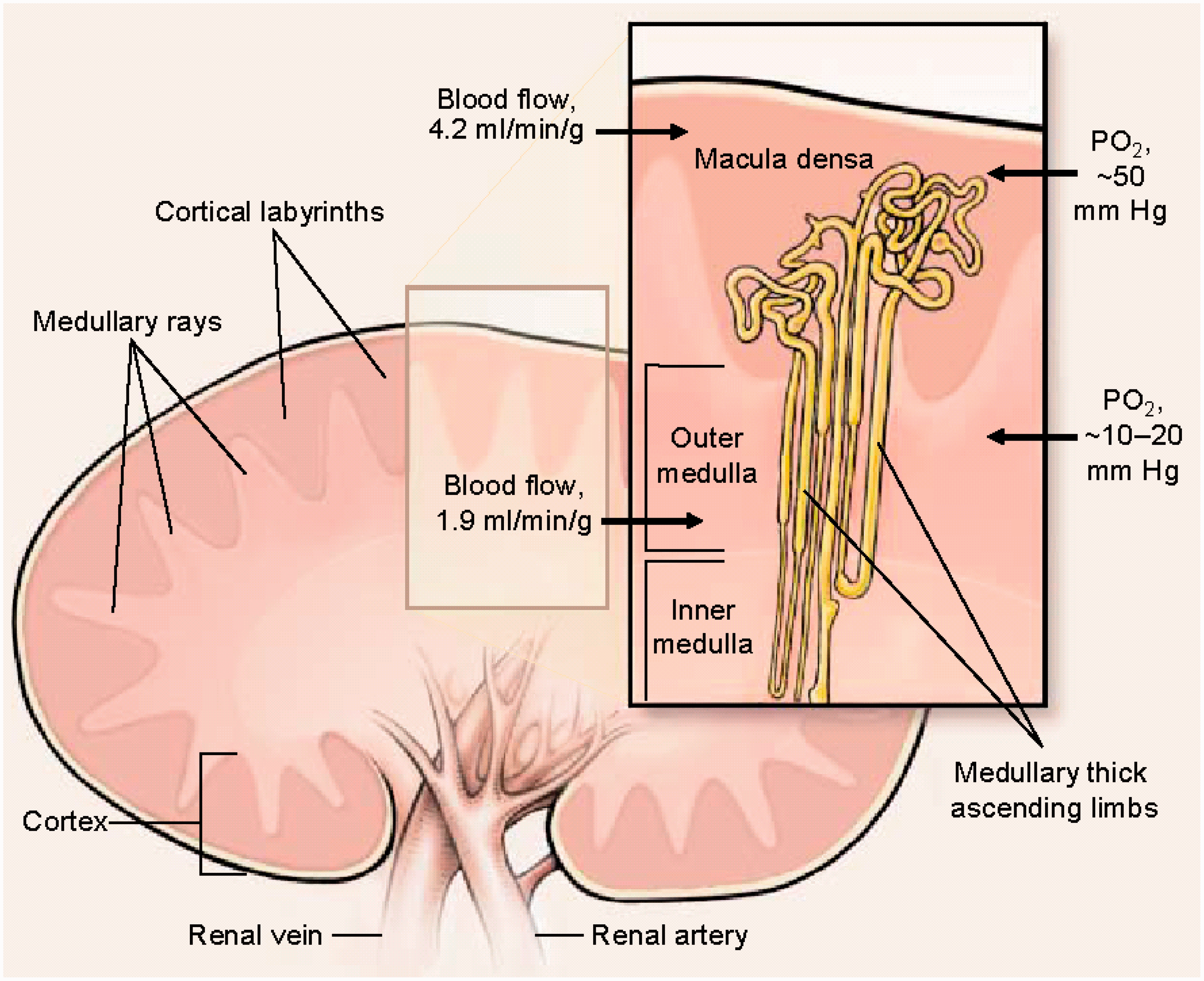
Other important haemodynamic and physiological factors involved in AKI pathophysiology are the regulation of glomerular blood flow (and the impact upon it of certain drugs), renal auto-regulation and tubulo-glomerular feedback.
Taking these in turn, the regulation of glomerular blood flow is important in maintaining GFR. Glomerular filtration is driven by the hydrostatic pressure within the glomerular capillary bed which, in turn, depends on the imbalance in blood flow into and out of the glomerulus (trans-capillary filtration pressure). The afferent arteriole is kept relatively vasodilated through the actions of eicosanoids, such as the prostaglandins, and nitric oxide; the efferent arteriole is kept relatively vascoconstricted through the actions of angiotensin II. Anything that reduces this imbalance, such as the administration of non-steroidal anti-inflammatory drugs (NSAIDs, which block prostaglandin production), or of renin-angiotensin system modifying drugs (such as angiotensin converting enzyme (ACE) inhibitors and angiotensin 2 receptor blockers (ARBs)), may reduce GFR. It is important to note that NSAIDs, ACE inhibitors and ARBs are rarely the sole cause of AKI but are frequent fellow travellers with other acute insults or with pre-existing risk factors such as chronic kidney disease.
The second factor is renal auto-regulation. As shown in Figure 3, GFR is normally tightly maintained over the physiological range of systemic mean arterial pressures (MAPs) through the actions of competing intrinsic renal vasoconstrictive and vasodilatory mechanisms. This relationship may, however, be disrupted by a wide variety of factors which means that GFR may start to drop even at MAPs that may be defined as ‘normal’ i.e. ‘normotensive’ AKI.
28
Table 3 carries detail of these different factors which can broadly be broken down into those that cause a failure to decrease arteriolar resistance, those that cause a failure to increase efferent arteriolar resistance and, finally, renal artery stenosis.
28
It is important to note that, once AKI is established, the renal auto-regulatory capacity (its ability to buffer the nephron from changes in systemic blood pressure) is lost; subtle changes in mean arterial pressure will be transmitted directly through to the renal microvasculature risking further injury and delayed recovery if insufficient attention is paid to maintaining renal perfusion – whilst avoiding ‘overshoot’ into hypervolaemia – in the ‘maintenance’ phase (Figure 1).
Normal and impaired auto-regulation of GFR as mean arterial pressure falls. Factors increasing susceptibility to renal hypoperfusion. Reproduced from Abuelo
28
with the permission of the Massachusetts Medical Society.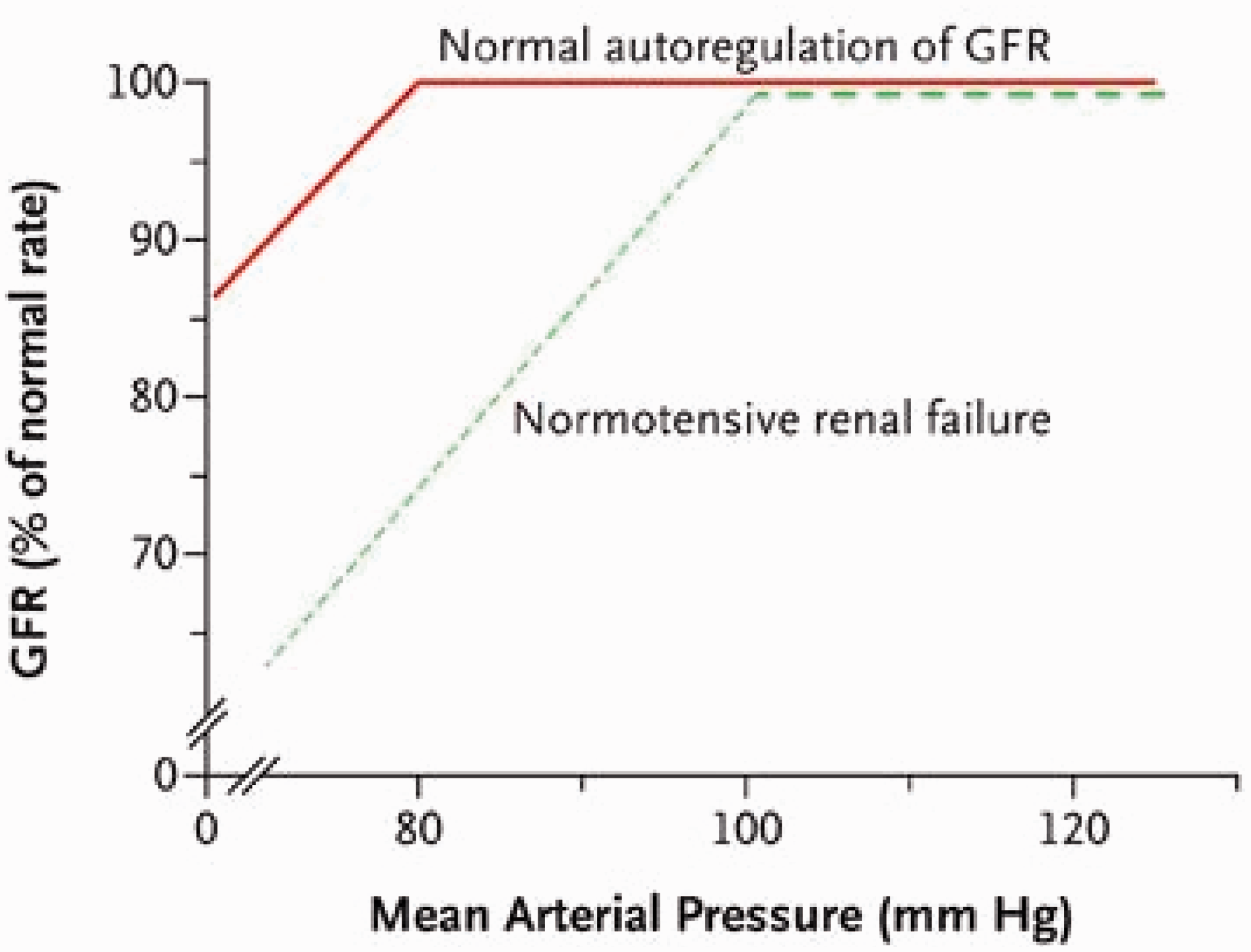

The third haemodynamic factor to discuss is tubulo-glomerular feedback which functions to regulate GFR and renal tubular flow. Increased delivery of sodium chloride to the distal tubule is detected by the macula densa, situated at the junction of the thick ascending limb of the loop of Henle and the distal convoluted tubule. This in turn causes a drop in GFR, which is thought to be mediated by an increase in afferent arteriolar resistance. 29 In AKI, activity of this negative feedback loop may be enhanced as a result of reduced re-absorptive capacity in the proximal tubule and loop of Henle, which, in turn, increases sodium chloride delivery to the macula densa and, ultimately, reduces GFR, further; vasoconstriction may be temporary, though, as there is some evidence that the mechanism can be ‘re-set’. 30
Endothelial injury
As shown in Figure 4 (adapted from Bonventre and Yang
24
), renal ischaemia-reperfusion injury leads to swelling of endothelial cells and disruption of the endothelial monolayer and the glycocalyx that overlies it.
31
These, in turn, cause a leak of fluid into surrounding tissues and interstitial oedema.
32
Endothelial cell injury. (a) Normal endothelium. (b) After ischaemia-reperfusion. 1: Endothelial cell swelling; 2: Disruption of the glycocalyx and endothelial monolayer, facilitating leucocyte trans-migration; 3: Back-leak of fluid leading to interstitial oedema; 4: Micro-thrombus formation; 5: Up-regulation of adhesion molecules (e.g. ICAM-1), enhancing …; 6: Leucocyte-endothelial cell interaction.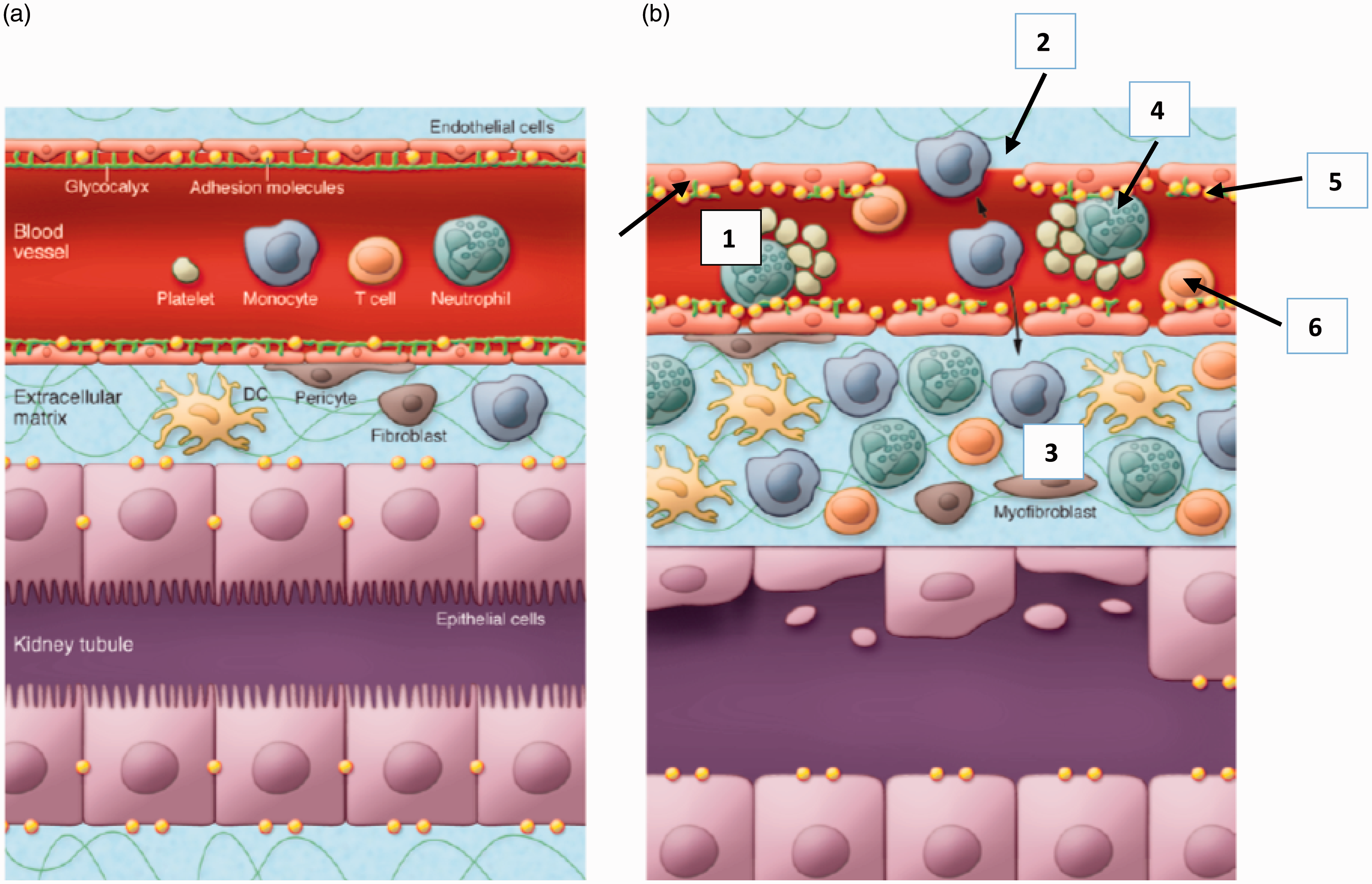
The initial ischaemic insult will be potentiated by a number of factors: activation of the clotting cascade, the formation of micro-thrombi within the vascular lumen and impaired vascular reactivity. Underlying the latter are a variety of factors including an altered balance between vasoconstriction, which is increased, and vasodilation, which is decreased; these seem to be explained by changes in small arterioles which become more sensitive to vasoconstrictors and less sensitive to vasodilators. 24 Reduced endothelial cell production of vasodilators, such as nitric oxide, contributes to the increased sensitivity to vasoconstrictors. 33
Endothelial cell injury also stimulates the inflammatory response that is so important in the pathogenesis of ischaemic acute kidney injury; up-regulation of various cell membrane adhesion molecules (e.g. inter-cellular adhesion molecule-1 (ICAM-1) 34 ) on the endoluminal surfaces of injured cells produces, via leucocyte-endothelial cell interaction, cytokine release (with further endothelial injury), micro-vascular luminal occlusion and leucocyte migration into the interstitium.
Vasoconstriction is compounded by the generation of various vasoactive cytokines (e.g. tumour necrosis factor-α (TNF-α) and a range of interleukins), as well as endothelin, as a consequence of leucocyte-endothelial interaction. 24
Epithelial cell injury
Although histological changes may be minimal (discussed in more detail, below), characteristic features include effacement and loss of the tubular brush border, sloughing of tubular epithelial cells into the tubular lumen, the development of areas of tubular luminal dilatation, the formation of tubular casts, and, where recovery has started, areas of cellular regeneration.35,36 Some of these changes are illustrated (Figure 5).
Tubular changes in ischaemic AKI.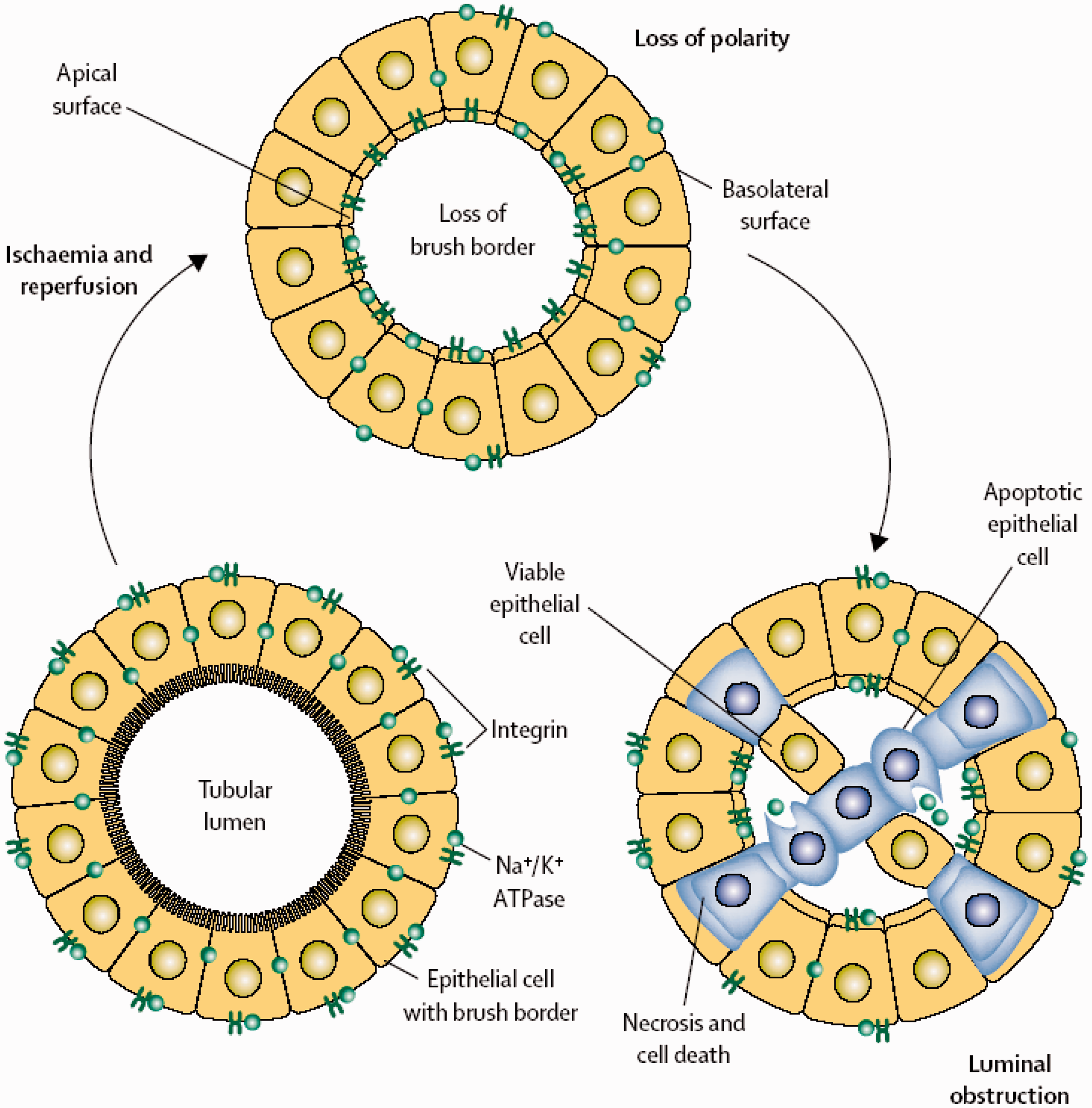
Histological changes seem to occur most frequently in the S3 segment of the proximal tubule (just before the start of the loop of Henle and within the outer medulla) in experimental models. 37 Human studies using the proximal tubule injury marker, kidney injury molecule-1 (KIM-1), confirm significant involvement of this region of the nephron.38,39 The relative protection of the straight segment of the distal tubule, despite being situated in a similar position in the outer medulla to the S3 portion of the proximal tubule, may be explained by various factors, including the superior ability to convert from oxidative to glycolytic metabolism, as already discussed. 27
Disruption of cell–cell and cell-basement membrane interaction explains the loss of otherwise intact tubular cells into the tubular lumen. This occurs following stress-induced redistribution of beta integrins from the lateral and basal membrane surfaces to the luminal surface. 40 Tubular obstruction can result from the binding of sloughed cells via these redistributed integrins. Tubular casts can form when cellular debris combines with tubular proteins such as Tamm-Horsfall mucoprotein 41 and fibronectin, 42 compounding luminal obstruction.
The functional decline evident in clinical AKI, however, usually exceeds the severity of histological change. There are a wide range of potential explanations:
changes may be restricted to the outer medulla (the vulnerability of which has already been discussed) and so may not be picked up in pathological samples; blockage of a single collecting tubule will affect multiple nephrons that drain into it; rather than necrosis, apoptosis may be the dominant mode of injury; the impact of tubulo-glomerular feedback, which will enhance the fall in GFR; tubular back-leak through denuded tubular basement membrane; and sub-lethal acute cellular injury (discussed next).
Experimental work has demonstrated evidence of redistribution of Na+K+ATPase from the baso-lateral to luminal membrane of the epithelial cell as a result of disruption of its actin cytoskeleton. 35 The resulting loss of cell polarity (and, thus, loss of cell function) may explain the ‘functional’ ischaemic AKI that can be seen in the absence of overt cell death and why (presumably as a result of restoration of cell polarity) this can recover in a much shorter time frame than might be expected when regeneration of a necrosed/apoptotic tubular epithelium needs to occur.
Experimental work has also shown redistribution of the complement receptor 1-related protein y (crry) complement inhibitor from the baso-lateral membrane, where it is constitutively expressed, allowing activation of the alternative complement pathway. 43 Unique to rodents, it is not clear if redistribution of analogous cell surface complement inhibitors occurs in human ischaemic AKI.
A variety of other cell surface receptors have been implicated in the injury phase of ischaemic AKI.
Toll-like receptors (TLRs), for instance, have generated interest recently as a potential therapeutic target. TLRs are a type of pattern recognition receptor (PRR) expressed in immune cells and other cell types, including the renal tubular epithelium. They play a key role in activating the innate immune response upon recognizing evolutionarily conserved microbial ligands (pathogen-associated molecular patterns, ‘PAMPs’), 44 such as bacterial glycolipids and lipopolysaccharide. Certain TLRs also recognize the so-called ‘alarmins’ (also known as damage-associated molecular pattern molecules, ‘DAMPs’) which are intracellular proteins liberated from stressed and necrotic cells. 45 TLR 2 and TLR 4, for instance, seem to play a pivotal role in the stimulation of the innate immune response to renal ischaemia-reperfusion injury (see also, below). 46
Finally, the proximal tubular epithelium promotes the immune response to ischaemia-reperfusion by also expressing major histocompatibility complex (MHC) class II molecules 47 (allowing antigen presentation to CD4+ T helper cells) and T cell co-stimulatory molecules. 48
Immunological factors
As discussed, damaged and stressed tubular epithelial cells play an integral part in potentiating the immune response to injury, resulting in the release of pro-inflammatory cytokines and chemokines and recruitment and activation of immune cells.
Both the innate and adaptive immune responses are involved in the pathophysiology of AKI.
The innate immune response is non-antigen-specific, occurring as early as 30 minutes after ischaemia-reperfusion, and includes direct injury to tubular epithelial cells by neutrophils, macrophages and natural killer cells, as well as complement activation (see below). Dendritic cells potentiate the innate immune response through the release of pro-inflammatory cytokines but also stimulate the adaptive immune response through antigen presentation. The adaptive immune response is antigen-specific, developing within hours of injury and lasting for a number of days. It includes maturation and antigen presentation by dendritic cells, T cell proliferation and activation and interactions between T and B cells.
The first wave of the immune system’s cellular response involves neutrophil recruitment which has a predilection for the outer medulla as it is the site of greatest injury. 49 The initial interaction with the endothelium (see also under ‘Endothelial injury’, above) causes the release of a variety of products such as myeloperoxidases and proteases, reactive oxygen species and cytokines, resulting in an increase in vascular permeability, cell injury and further leucocyte recruitment. 24
The second wave of the immune system’s cellular response involves the recruitment of circulating blood monocytes and their differentiation into macrophages, apparent as early as 1 h after experimental ischaemia reperfusion and appearing to persist for around seven days after an early peak at 24 h. 50 Macrophages’ chief function is the phagocytosis and digestion of cellular debris as well as pathogens but also play a key role in the adaptive immune response through antigen presentation. Macrophages of the M1 phenotype have a dominant “killer” function and partner the Th1 (T helper cell type 1) cellular immune response. M2 phenotype macrophages, in contrast, are involved in the Th2 humoral immune response but seem also to have a role in tissue repair. 51
Finally, in terms of the immune cellular response, T lymphocyte infiltration of damaged renal tissue is evident as both an early and subsequently persisting response after ischaemia-reperfusion with CD4+ cells implicated in propagating injury. 52
Complement activation plays a key role in the innate immune response with experimental evidence of local synthesis of C3 within hours of experimental ischaemia reperfusion. 53 Unusually, it is the alternative complement pathway that seems to be exclusively activated in the setting of ischaemic AKI. 54 Complement activation has a number of effects including chemokine production by the tubular epithelium 55 and direct cell injury through the actions of the membrane attack complex (C5b-9). 56
The importance of redistribution of the rodent-specific crry cell surface complement inhibitor in allowing activation of the alternative complement pathway 43 has already been discussed (under ‘Epithelial cell injury’). Indeed, it has been shown that heterozygous, gene-targeted mice expressing lower concentrations of crry are more vulnerable to ischaemic injury. 43 The relevance to complement activation in human ischaemic AKI is uncertain.
Sepsis
To complete this section of the discussion, it is worthwhile considering the specific circumstance of sepsis as a cause of AKI, its commonest aetiology in the critically ill.
The evidence of what happens to renal blood flow in sepsis gives no clear answer. The haemodynamic response in humans is uncertain 57 and whilst experimental work reports that overall flow may be unchanged, increased or decreased, some evidence suggests that septic AKI is a hyperaemic phenomenon with the generalized vascular dilatation characteristic of sepsis upsetting regional glomerular haemodynamics and the trans-capillary filtration pressure, 58 even though whole organ flow may be preserved.
Other evidence suggests that the explanation may not be quite so straightforward. Sepsis is characterized by nitric oxide-induced arterial vasodilation with a consequent drop in systemic vascular resistance. 21 The neuro-humoral response to arterial under-filling includes activation of the sympathetic nervous system, the renin-angiotensin-aldosterone axis and non-osmotic release of vasopressin, but these mechanisms may contribute to AKI by causing renal vasoconstriction. 21 Renal vasoconstriction may be further compounded by tumour necrosis factor α-induced release of endothelin. 59 Haemodynamic support using noradrenaline may also affect renal function by increasing afferent arteriolar resistance, thereby reducing GFR (vasopressin may cause efferent arteriolar vasoconstriction and so may actually increase GFR). 21
Vasodilatory mechanisms that might serve to counter renal vasoconstriction may be attenuated; experimental work has suggested that increased plasma nitric oxide concentrations arising from endotoxaemia-mediated increases in inducible nitric oxide synthase (NOS) may actually down-regulate endothelial (constitutive) NOS within the kidney. 60
Histological changes in sepsis-associated AKI may be minimal or absent, 61 although clinically (in terms of natural history) septic AKI seems to ‘behave’ in much the same way as other causes of ischaemic AKI. Biochemically (in terms of loss of sodium re-absorptive capacity) septic and non-septic ischaemic AKI also seem to behave similarly, at least in experimental models; 62 the picture is less clear in humans although the evidence base is far from robust. 63
Recovery
Assuming the patient survives the acute illness, recovery processes are initiated soon after injury and involve the actions of endogenous inhibitors of inflammation, the up-regulation of a variety of different repair genes, the actions of various cellular components of the immune system, the clearance of debris and tubular regeneration.
Various endogenous inhibitors may be important in limiting inflammation, for instance, haem-oxygenase 1, whose production in tubular epithelial cells is increased after ischaemia, 64 and Tamm-Horsfall muco-protein, a normal tubular secretory product which may also down-regulate expression of cell surface TLR 4 in outer medullary proximal tubular cells. 65
The recovery process includes the up-regulation of a variety of different genes, such as those for KIM-1 in the proximal tubule and neutrophil gelatinase-associated lipocalin (NGAL). KIM-1 is involved in the clearance of apoptotic and necrotic cells by the proximal tubule. 66 NGAL, produced in the distal tubule, is circulated, filtered by the glomerulus and then reabsorbed by the proximal tubule where it seems, in experimental models, to inhibit apoptosis and induce cellular proliferation. 67 Its protective role may occur by facilitating iron sequestration, preventing the formation of reactive oxygen species. 67 The appearance of these products arising from ischaemia-reperfusion injury has shown some promise as early diagnostic evidence of AKI in clinical research, but their precise role in clinical practice remains unclear. 68
Another important component of the recovery process is the clearance of necrotic and apoptotic cells. This is mediated via macrophages, but also by intact tubular epithelial cells, themselves. The phagocytic properties conferred through the actions of KIM-1 66 allows such cells to recognize a specific surface epitope expressed on apoptotic cells. The clearance of apoptotic cells may help limit the inflammatory response. M2 macrophages (see above) seem to predominate in damaged tissue after 3–5 days when repair is already underway. 69 In addition, a specific subset of dendritic cells may also be involved in tissue regeneration 70 as may a certain sub-type of regulatory anti-inflammatory T cells that infiltrate the kidney 3–10 days after experimental ischaemia reperfusion. 71
Recent years have brought controversy about the provenance of cells re-populating the tubular epithelium with potential candidates including surviving tubular epithelial cells, bone marrow stromal cells and renal progenitor cells. It seems, though, that although bone marrow stromal cells do migrate to the damaged epithelium, they function to exert a paracrine, anti-inflammatory action72,73 and that it is surviving tubular cells that re-populate the epithelium. 74
Abnormal repair and recovery
The received clinical wisdom had been that, provided the patient with AKI survived, full renal recovery usually ensued. Increasing evidence suggests, though, that this is not the case7–9 and that subtle pathophysiological changes contribute to increased long-term renal attrition after the acute phase of illness. The immediate clinical consequences of this include persisting renal dysfunction, which may progress to end-stage kidney disease (ESKD).8,9 There is also an association with reduced long-term survival even in those without ESKD8,9 and whether this relates to sequelae of CKD, such as hypertension, or reflects higher vascular mortality in those who are prone (by dint of their vascular disease) both to AKI and to attenuated recovery post-AKI, is not clear. Given the importance of these longer term sequelae, though, it is worth understanding some of the underlying mechanisms contributing to abnormal repair.
The clinical end-point of abnormal repair is chronic kidney disease which is reflected, histologically, by tubular atrophy and renal fibrosis, due to myofibroblast proliferation and deposition of extra-cellular matrix. There appear to be three routes to these end-points.
First, there may be a non-recoverable loss of the capillary network. There appears to be a reduction in the density of the peri-tubular microvasculature after ischaemia-reperfusion injury. 31 This impaired new vessel formation may arise from reduced production of vascular endothelial growth factors (VEGF) and up-regulation of inhibitors of angiogenesis. 75 The resultant chronic hypoxia leads to fibrosis which, itself, by interfering with the local micro-vasculature, exacerbates hypoxia and promotes further progressive fibrosis 76 and the clinical end-points of chronic kidney disease and hypertension.
Second, a chronic activation of the immune response, particularly of macrophages, may persist leading to the release of pro-fibrotic cytokines such as transforming growth factor – β1 (TGF-β1). 77
Finally, proliferating tubular epithelial cells may undergo cell cycle arrest at the G2/M interface leading to the production of TGF- β1 and connective tissue growth factor. 78 Epigenetic modifications may cause persisting fibroblast activation and fibrosis. 79
Mortality correlates
As already discussed, AKI of only modest severity is associated with an increased mortality 4 but still appears to be an independent risk factor for death rather than a fellow traveller with other acute co-morbidity. 5 One cannot easily cite the ‘traditional’ causes of death due to advanced renal failure such as the uraemic syndrome, pulmonary oedema or hyperkalaemia in these circumstances. Recent developments in our understanding of the remote effects of renal ischaemia-reperfusion injury provide more intriguing insights into the dangers of AKI – so called ‘organ cross talk’.
Kelly and colleagues, for instance, demonstrated a rapid increase in systemic TNF-α concentrations in experimental animals with induced renal ischaemia but not in controls (sham operation but with no renal vascular ligation). 80 These increases were sustained over at least 24 h. Most striking, though, were the remote effects on the heart with increased cardiac myocyte expression of ICAM-1, inflammatory cell infiltrates within myocardial tissue and evidence of myocyte apoptosis. These cellular-level changes were reflected, functionally, by changes in cardiac contractility. Importantly, these remote effects were not seen in nephrectomized controls, i.e. this seemed not to be a consequence of the accumulation of uraemic toxins, per se, but of the inflammatory consequences of ischaemia-reperfusion injury.
Other experimental work gives insights into the particularly dangerous association of ischaemic AKI with increased lung water; down-regulation of sodium and water channels in the lung was evident after bilateral ischaemia-reperfusion injury but not in sham controls 81 offering a potential explanation for impaired lung water clearance. In this work, however, uraemic toxins rather than inflammatory products were cited as causative as bilaterally nephrectomized animals – but not those with single kidney ischaemia-reperfusion injury – showed similar lung changes.
Conclusions
In this review, we have seen how disordered renal auto-regulation may explain the higher risks of ischaemic acute kidney injury in those patients with chronic kidney disease and other conditions and in those taking non-steroidal anti-inflammatory drugs, ACE inhibitors and angiotensin 2 receptor blockers.
We have also seen that ischaemic AKI is an intensely pro-inflammatory state. The complex interplay of endothelial and epithelial cells with the immune system potentiates the damage caused by the initiating renal insult.
Chronic kidney disease is an increasingly well-recognized sequela of AKI and may be explained by maladaptive repair.
Finally, AKI may exert remote effects on other organs. This organ cross talk may help explain the increased mortality associated with the condition even at relatively modest concentrations of renal dysfunction.
Footnotes
Acknowledgements
This article was prepared at the invitation of the Clinical Sciences Reviews Committee of the Association for Clinical Biochemistry and Laboratory Medicine.
Declaration of conflicting interests
None declared.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethical approval
Not applicable.
Guarantor
NSK.
Contributorship
NSK is the sole author.