
Abstract
‘Measurement uncertainty of measured quantity values’ (ISO15189) requires that the laboratory shall determine the measurement uncertainty for procedures used to report measured quantity values on patients’ samples. Where we have numeric data measurement uncertainty can be expressed as the standard deviation or as the co-efficient of variation. However, in immunology many of the assays are reported either as semi-quantitative (i.e. an antibody titre) or qualitative (positive or negative) results. In the latter context, measuring uncertainty is considerably more difficult. There are, however, strategies which can allow us to minimise uncertainty. A number of parameters can contribute to making measurements uncertain. These include bias, precision, standard uncertainty (expressed as standard deviation or coefficient of variation), sensitivity, specificity, repeatability, reproducibility and verification. Closely linked to these are traceability and standardisation. In this article we explore the challenges presented to immunology with regard to measurement uncertainty. Many of these challenges apply equally to other disciplines working with qualitative or semi-quantitative data.
Introduction
‘“That’s right”, shouted Vroomfondel, “We demand rigidly defined areas of doubt and uncertainty!”’ Douglas Adams, Hitchhiker’s Guide to the Galaxy, 1979.
Definitions of uncertainty are not all helpful but all imply an element of doubt caused by a situation in which something is not known. Measurement uncertainty can be quantified statistically where we have numerical data. However, in immunology many of the assays are reported either as semi-quantitative (i.e. an antibody titre) or qualitative (positive or negative) results. In the latter context, measuring uncertainty is considerably more difficult. There are, however, strategies which can allow us to minimise uncertainty. The approach advocated herein for a range of immunology assays addresses both sorts of data.
Measurement uncertainty
Measurement of uncertainty provides a quantitative assessment of the quality of a test result. The old Clinical Pathology Accreditation (CPA) standard advocated that we ‘determine the uncertainty of results … where possible’. 1 The current international standard (ISO 15189) 2 has a tighter requirement, instructing us to consider ‘measurement uncertainty of measured quantity values’. As noted earlier, in immunology we must also understand, and attempt to minimise, the measurement uncertainty of the many assays producing a purely qualitative result.
The concept of measurement uncertainty was originally devised for use in measurements in physics. The total error of a measurement is a combination of random error (measured by the standard deviation, SD) and systematic error (bias). The measurement uncertainty principle is that bias should be corrected for by reference to standards, leaving the SD as the basic parameter for measuring uncertainty. As we will see, the lack of standards makes this much harder to isolate as the sole source of error in the biological sciences.
Causes of measurement uncertainty can occur at any stage of the laboratory process, i.e. preanalytical, analytical or postanalytical. Preanalytical specimen requirements and collection criteria are defined by the laboratory. Users of the service should have access to the requirements through the local laboratory users’ guide. This should be reinforced through training and teaching, for example in induction sessions at the start of employment, and through user group interaction.
The bulk of our discussion focuses on the analytical phase. A number of parameters can contribute to making measurements uncertain. These include bias, precision, standard uncertainty (expressed as SD or coefficient of variation [CV]), sensitivity, specificity, repeatability, reproducibility and verification. Closely linked to these are traceability and standardisation.
Standardisation
Standardisation of assays for the determination of autoimmunity is an international problem and at present there is no unified international solution to resolve these problems. This is due to the fact that antibodies are difficult to define unequivocally due to their molecular structure, weight and biological variability and that they reside within a complex matrix (serum) and therefore cannot be directly measured. 3 Furthermore, the quantification of an autoantibody relies on its functional ability to bind antigen, with consequent variation between samples and standards of affinity, avidity and epitope specificity. In consequence, the uncertainty of measurement with these assays is high. Variability and therefore uncertainty may be introduced at many levels, including the antigen source (whole tissue, cell extract, purified protein, recombinant protein), antibody detected (isotype, affinity, concentration), antibody detection system (polyclonal, monoclonal, affinity, conjugation [enzyme, fluorochrome] and methodological variations [incubation time, volume, choice of substrate]). As a result we have to ensure the maximum adherence to quality control measures in order to minimise subsequent assay uncertainty.
Autoantibody standards
Examples of reference materials available for use in clinical immunology laboratories.

ANA: antinuclear antibody; CDC: Centers for Disease Control, Atlanta, USA; CRP: C-reactive protein; ENA: extractable nuclear antigen; IRMM: Institute for Reference Materials and Measurement, Belgium; NIBSC: National Institute of Biological Standards and Control, Potters Bar, UK; WHO: World Health Organisation.
DA470k, although now the international reference material for many proteins, including immunoglobulins and complement, is not generally available to diagnostic laboratories. Many commercial preparations (secondary standards) are available, calibrated against this material.
Protein standards
International calibration material is available for 12 serum proteins (ERM-DA470k/IFCC) including immunoglobulins G, A and M; complement components C3c and C4. Most, if not all, commercial companies derive their secondary commercial standards for specific proteins from ERM-DA470k/IFCC. This makes it possible for laboratories worldwide to compare these specific protein results over time and between hospitals and countries. Without such defined materials standardisation of assays is impossible as each manufacturer can define their own arbitrary target units.
Even with certified reference preparations harmonisation of results between manufacturers may not be achievable. A recent commutability study for caeruloplasmin, using the original and new reference preparations (ERM-DA470 and ERM-DA472/IFCC, respectively), failed to show standardisation for this measurand across multiple analytical platforms all of which claimed to be traceable to the same calibration preparation (ERM-DA470). 5
Traceability
For many autoantibody methods there is limited traceability of the standards, going only as far as manufacturers’ in-house selected measurement procedures or master calibrators. Often these are assigned an arbitrary numeric value. Users should be advised that results are not directly comparable with results generated by different measurement procedures. 3 There are numerous examples of divergence in the numeric values assigned to quantified autoantibody tests (e.g. antitissue transglutaminase, 6 antithyroid peroxidase 7 ). In the case of antithyroid peroxidase the two antigen sources (purified or recombinant) differ in glycosylation and are likely to express different conformational epitopes. In some patients this leads to very different binding of the autoantibodies, resulting in very different quantified results.
Even where there is good traceability there is often no agreed standard. There is currently much interest in IgG4 quantification in IgG4-related disease. 8 However, there is no internationally agreed standard. The two main methods used in the UK (The Binding Site Group Ltd. and Siemens Healthcare Diagnostics) are traceable to two different standards. The Binding Site calibration material is traceable to the CRM470 whereas the Siemens calibration material is traceable to WHO67/97.9,10 This results in a situation where the IgG4 results from the Siemens assay are twice those of the Binding Site assay. 11
Methodological standardisation
Published consensus documents exist for the use of some assays but most of these only relate to appropriate use of generic tests for disease diagnosis/monitoring rather than prescription of which analytical system to use (e.g. ESPGHAN (European Society for Pediatric Gastroenterology, Hepatology and Nutrition) for coeliac disease, 12 NICE (National Institute for Health and Clinical Excellence) guidance for coeliac disease 13 and Rheumatoid arthritis 14 and international consensus document of antineutrophil cytoplasm antibody (ANCA) testing and reporting 15 ).
Having a good understanding of the analytical aspects of assays, including knowledge of the biology of the antibodies that we are detecting and of the clinical significance of results, is imperative in ensuring that results are given the correct interpretation. In particular, the user should appreciate the difficulty in controlling such variables as those introduced by changes in the antigen (e.g. animal or human, purified or recombinant), the sensitivity of the detection system (e.g. for rheumatoid factor agglutination (coated cells, gelatin beads or latex), ELISA (enzyme-linked immunosorbent assay) or fluorescence immunoassay) and the antibody mixture in the serum being analysed (avidity, affinity, antibody class (IgG, IgA, IgM or a combination of these) and epitope specificity).
Applications in clinical immunology
Immunology assays fall into three categories: qualitative assays, semi-quantitative assays and quantitative assays. Each will be discussed with reference to what factors make the results uncertain.
Qualitative assays
Qualitative assays include all IIF tests (e.g. antinuclear antibody (ANA), ANCA). The principle of these assays and the microscopic appearance of a typical assay are shown in Figure 1. These are subjective tests and therefore by their nature likely to have a high degree of uncertainty. Variability of reagents can be down to choice including the substrate (e.g. rodent tissue versus human cell lines for ANA), fixatives (e.g. pure ethanol or ethanol plus proprietary additives for ANCA), serum dilution, conjugation (fluorescence versus enzyme) and the quality of the secondary antibody (affinity, avidity, antibody class detected). Even the setup of UV (ultraviolet) microscopes (mercury versus light-emitting diode light sources, magnification, numerical aperture) will have an impact on the results obtained.
Whilst reagents are available as kits for some assays, for many we have to select individual components, each of which must be optimised to maximise the sensitivity and specificity, for example by altering sample or reagent concentrations or changing incubation times. The laboratory may be directed by the manufacturer as to what is optimal for the autoantibody of interest (e.g. a 1 in 20 initial dilution of serum for multiple autoantibody screening on rodent liver/kidney/stomach composite sections [NOVALite, INOVA Diagnostics, Inc., San Diego, USA]). Determination of positivity/negativity is set based on the use of controls as well as operator consensus opinion and/or expert operator judgement.
(a) Typical design for indirect immunofluorescence assays. (b) Appearance of antinuclear antibody on Hep2 cells (speckled pattern; nucleoli are spared; no cytoplasmic stain). (c) Appearance of antinuclear antibody on Hep2000 cells (speckled pattern variable owing to overexpression of Ro60 in some cells; nucleoli variable; no cytoplasmic stain).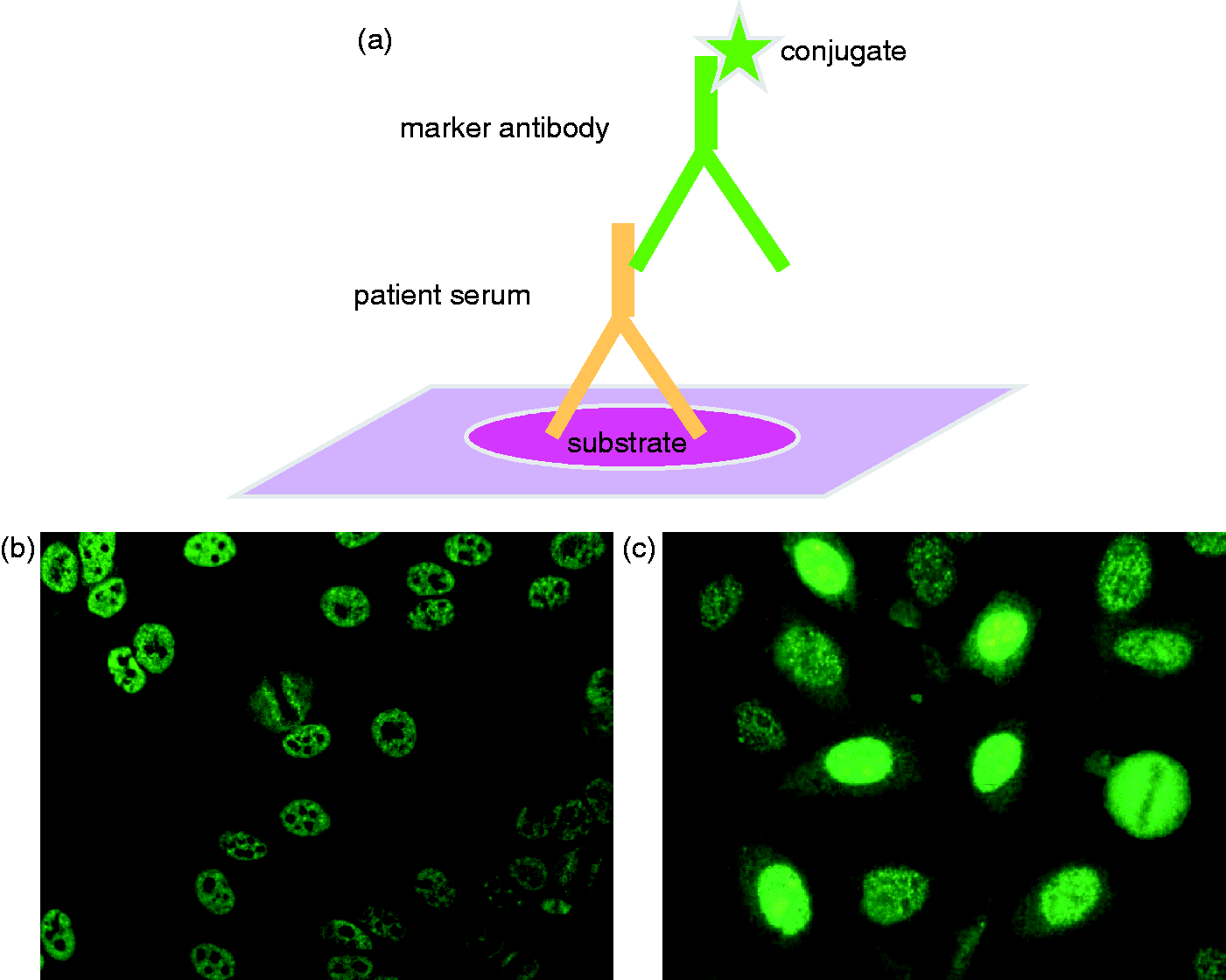
Qualitative assays – substrate choice and uncertainty
As there is no standardisation of substrate preparation, differences between manufacturers can be seen. Substrate preparation can vary in choice of cell line and whether they are transfected with additional antigens, for example use of HEp2 cells versus HEp2000 for ANA determination. HEp 2000 cells are transfected with, and hyper-express, SS-A/Ro60 antigens for ANA detection (Figure 1(b) and (c)). Potential variations in tissue section preparation include cross-section of tissue, orientation, fixation and whether multiple tissues are arranged as a composite block, i.e. subsections of individual wells (e.g. rodent liver, kidney and stomach slides). What is deemed best for an individual lab may depend on laboratory and clinical expertise and also personal preference. In this context it is worth noting that rodent tissue is relatively poor in Ro60 antigen and some patient serum samples with only this specificity may be negative on rodent composite blocks.
Semi-quantitative assays – substrate choice and uncertainty
Semi-quantitative assays include immunoblot methods. In these assays, substrates can be recombinant and/or purified antigens which are bound to nitrocellulose or other membranes. The most common commercial format has a series of antigens of interest immobilised on a single strip. Examples would include panels for myositis-related antibodies (e.g. Jo-1, PL7, PL12, OJ, EJ, etc.) (e.g. Euroline Myositis profiles, Euroimmun, Luebeck, Germany) or for paraneoplastic antibodies (e.g. Purkinje cell, antineuronal nuclear antibodies 1 & 2, amphiphysin, Ma2) (e.g. Euroline neuronal antigen profile, Euroimmun, Luebeck, Germany). There is no standardisation of antigens for these assays and the sensitivity and specificity vary accordingly. Reagents are usually produced as a kit and therefore have a manufacturer validated methodology. In this context (ISO15189), 2 a validated assay is one for which extensive studies have been undertaken to provide objective evidence of the performance characteristics of the assay (including, for example accuracy, precision, sensitivity and specificity). Note that it is still incumbent upon the user of the assay to verify the manufacturer’s claims and this may require a significant resource input in terms of both time and reagents. All immunoblots include positive and negative controls on each membrane to authenticate each test.
ELISA methods that generate a ratio result are also categorised as semi-quantitative. Again, there is great variation in the substrates used. Screening ELISAs such as those used for ANA or extractable nuclear antigen (ENA) detection may contain a variety of purified or recombinant antigens, the exact combination of which will vary from manufacturer to manufacturer. These assays utilise a single calibrator (set by manufacturer), the optical density (OD) of which is multiplied by a factor (given by the manufacturer specific to each lot number) to give an OD cut-off by which all patient ODs must be divided to generate a ratio.
Other examples of semi-quantitative assays in immunology include assays for functional components of the immune system. These include complement (classical pathway CH50, alternate pathway), assays for neutrophil function and lymphocyte proliferation. Again, these rely largely on single point calibration, usually a fresh random normal sample.
Quantitative assays
Quantitative assays include all ELISA-based autoimmunity and allergy tests (including those that are measured as quantitative but may only be reported as qualitative, e.g. ENA profile assays). Some examples of quantitative assays, and their associated limitations, are discussed below.
Substrate choice and uncertainty
Source of antigen was noted earlier as being one area for substantial variation between assays. For a few assays (e.g. cyclic citrullinated peptide) the patented antigen is supplied from a single source under licence to other manufacturers. Thus, differences between manufacturers can be reduced due to the limited variation in antigen preparations available. Yet even here variation has been introduced at a later point, with some manufacturers using other citrullinated antigens (e.g. Mutated citrullinated vimentin) thereby bypassing the patent issue. Whilst clinically equally valid these two antigen sources identify overlapping but different patient populations. 16 The analytical differences between these assays are now being reflected in the UK NEQAS (National External Quality Assessment Service) scheme for anticitrullinated protein antibodies. 17
Variability of substrate is even greater for other autoantigens. We offer one example. Anticardiolipin antibodies, as seen in the antiphospholipid syndrome, rely on the binding of a co-factor, beta-2-glycoprotein-1(βGP1), which results in a neo-antigen to which the antibodies in turn bind. Current guidelines require the addition of βGP1 either bovine or human and that this may be by addition of purified human protein or bovine serum. 18 The type of solid phase, concentration of antigen and method of coating are also recognised to contribute to the variation in substrate but are not addressed by the guideline.
Standards and uncertainty of autoantibody assays
ELISA-based assays present a challenge as there are few international standards for autoantibody preparations. Examples would include the ‘Harris standards’ (three generations have been available from Louisville APL Diagnostics, US traceable to the original Harris standards), the original anticardiolipin calibrators which were samples containing specified concentrations of antibodies 19 and the WHO designated serum Wo80 for anti-dsDNA (Wo80) which unfortunately is no longer available. As a result, there is potentially huge assay uncertainty which is shown by the variability of dynamic ranges and positive cut-off values between manufacturers for the same assay, as seen in many external quality assessment (EQA) returns in immunology. This is noted even in assays with an international reference preparation (e.g. anticardiolipin17,20).
This is further highlighted by EQA schemes where most only require qualitative responses for some assays which are routinely reported quantitatively or semi-quantitatively. For example, for quantified anticardiolipin antibodies the interlaboratory CV for is often high (>25%). 21 In a report on UK NEQAS data in 1995 the authors concluded ‘On the basis of the evidence there seems little room for confidence in the concordance of numeric values for cardiolipin antibodies, and only slightly more confidence in their semiquantitative interpretation’. With the slow progress in agreement on international standards, there is little to suggest that this situation has improved in the intervening two decades. Changing to a qualitative approach achieved higher interlaboratory concordance than the numeric data in the UK NEQAS phospholipid scheme. 4
Quantitative autoantibody assays are measured as IgG, IgA or IgM autoantibodies depending upon the isotype of clinical interest. Some assay systems (e.g. Phadia ImmunoCAP 250 EliA, Thermo Fisher Scientific Inc., Uppsala, Sweden) use immunoglobulin calibrators which are traceable via an unbroken chain of calibrations to the International Reference Preparation (IRP) 67/86 of Human Serum Immunoglobulins A, G and M from the WHO; measured in µg/L. A lot specific correction factor is applied to each batch of autoantibody specific assay wells and is used to automatically convert the mass unit results (μg/L) to the arbitrary units (e.g. U/mL) that are reported. All autoantibodies are measured in arbitrary units (U/mL or IU/mL) as there are no equivalents of SI units. Use of such correction factors must rely on manufacturer specific criteria to be appropriately set. Clearly, this does not always work effectively. Examination of EQA data even for the measurement of serum proteins shows some manufacturers to have introduced reproducible and consistent bias. Variability between lots may result in variability of correction factors which potentially may affect end results generated. This may be highlighted in lot specific shifts observed in cumulative internal quality control (IQC) results.
Owing to the lack of standardisation in autoantibody measurement, different manufacturers utilise different arbitrary units and positive cut-off values. Positive cut-offs provided by manufactures ought to have a good evidence base but should be verified by laboratories. Whist it is advised to adjust/set up in-house reference ranges based on local population cohorts this is often not feasible particularly in a district general hospital setting. Limited patient numbers and the ethical issues of large normal control studies may mean that small cohort studies would lead in inaccurate results. For many ‘data mining’ is one feasible way to overcome this. Data mining, also called knowledge discovery, is a term borrowed from computer science whereby raw data in large databases is turned into useful information. One example of this applied to immunology is the use of probability plots to isolate normal from abnormal populations within a database. 22
Standards and uncertainty of assays for allergy testing allergen specific IgE and total IgE
Total serum IgE concentrations are reported in international units or nanograms per millilitre (1 IU/mL = 2.44 μg/L). Total IgE is cross-standardised with the WHO 75/502 human reference IgE serum verified by periodic proficiency surveys. 23 As with total IgE, some commercial specific IgE antibody assays are calibrated using heterologous interpolation against the WHO 75/502 human IgE reference serum, thereby enabling a uniform system of reporting as international units of IgE per millilitre. This value can be converted into mass units of IgE per volume (e.g., micrograms of IgE per litre) because the IgE calibration curve is standardised against the WHO Human IgE International Reference Preparation 75/502. Note that this ignores the problem that to measure total IgE we use reagents that react with the constant region of the IgE molecule (Fc, fragment crystallisable), giving us some hope of measurement in mass units. In contrast, specific IgE testing relies on the active binding site of the patient’s IgE (located in the Fab region [fragment antibody binding]) which will differ in epitope specificity and affinity/avidity from patient to patient. This must have an effect on the amount of specific IgE bound in a given system. In addition, source and preparation of allergen (purified, recombinant) is chosen by the manufacturers which will introduce further variation. Fortunately, the small number of manufacturers delivering specific IgE testing (five represented on UKNEQAS) should limit the problem.
Total serum mast cell tryptase concentrations are reported in μg/L. The UK NEQAS for Tryptase EQA scheme shows all participants utilise the Phadia ImmunoCAP methodology albeit on the various platforms available. Calibrators are calibrated against a manufacturer’s internal tryptase reference material, which has been purified from human lung according to the protocol published by Schwartz. 24 As all three platforms and their reagents are manufactured by the same supplier the preparation of the reference material across platforms should be standardised. However, it is clear that, despite there being a single supplier, there is significant bias according to the platform used. 17 Nevertheless between laboratory reproducibility, which should be similar to ELISA (between batch variation 8–12% 4 ), is acceptable at around 10%. 17
Analytical controls to reduce risk
Given all the analytical variability described earlier, how can we ensure that our immunoassays are generating results as accurately and reliably as possible? Below are examples of what we believe to be necessary actions that should be applied to the different methodologies. While the examples given are from immunology, much of the advice could be equally well applied to other laboratory disciplines.
For qualitative assays
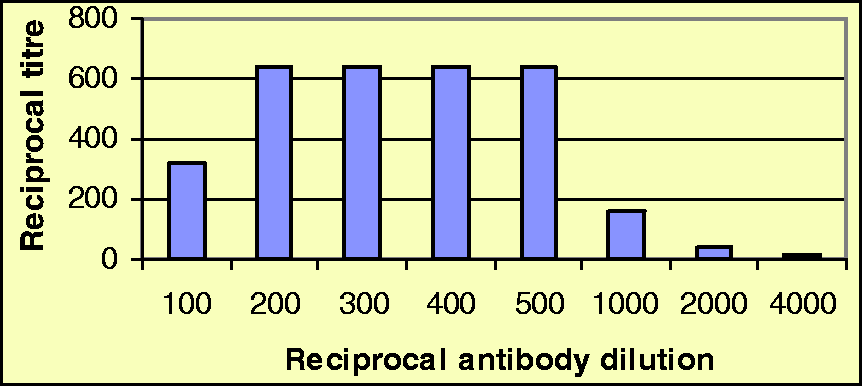
Validation methods include confirmation of clinical sensitivity and specificity where possible. This requires a significant input of resources in terms of reagents and time (both scientific and clinical) and may not be achievable in all centres. In this case it is essential that the laboratory thoroughly research the literature to understand how the assay performs. Comparison between manufacturers may be necessary. Indeed, this is vital when replacing one assay with another in the laboratory’s repertoire. This is essential if the end users are to be aware of the impact of the change on their patients, either at diagnosis or for monitoring response to therapy. Analytical tools and appropriate statistical evaluation for this are limited for qualitative assays but can be applied more extensively for quantitative assays. Use of IQC material. Positive and negative serum samples should be included in each batch. Sensitivity can be monitored by including a positive sample in serial dilutions (usually twofold serial dilutions) to give a titre. The end result should be plus or minus one increment (e.g. if the expected titre is 1 in 320 the acceptable range would be 1 in 160 to 1 in 640). Enrolment in EQA schemes, where they exist. CPA accreditation
1
requires that where no such schemes exist, alternative mechanisms of comparisons should be employed such as sample exchange arrangements. Clinical authorisation with appropriate clinical commentary. Note that full interpretation would require knowledge of the positive and negative predictive values of the assay in the context of your local population. Use of CE (Conformité Européene) marked reagents not necessarily as part of a standard kit. This requires regular re-evaluation, e.g. of appropriate dilution of fluorescein isothiocyanate-conjugated antisera. If the antiserum is too concentrated one risks a prozone effect (reduced stain, the mechanism of which is uncertain), wastes money and potentially increases background stain. If too dilute there is a loss of sensitivity. The standard approach is to use a known positive serum of defined titre to create a checkerboard with the conjugated antiserum. The appropriate dilution is the last but one before the fall off in sensitivity. This allows long-term storage without risking a drop in sensitivity. An example of this is shown in Figure 2. Expertise in IIF reading. Audit of IIF reading with comparison of user agreement and regular revalidation of competency exercises. Practically, this first requires the collection of a panel of serum samples with defined specificities (for example a panel for antinuclear antibodies would include homogenous, speckled, nucleolar, centromere, mitotic spindle apparatus, etc.). These are then used to create a test slide which should be assessed ‘blind’ by all staff involved in the regular reading of IIF. Similar panels may be created for other specificities, e.g. ANCA, skin antibodies. Definition of ‘correct result’ must be established whether this be based on an expert opinion or based on consensus. Where any reader is considered out of consensus then re-education is the key to quality improvement. Regular maintenance of analysers, diluters and UV microscope. For semi-quantitative assays
As above a–d and h plus. Use of CE marked commercial kits. It is possible to use non-CE marked kits provided that adequate evidence of assay maintenance is available, however this may be prohibitive for most routine laboratories. For quantitative assays
As above plus. Use of manufacturer validated reference ranges, internally verified as appropriate. Scrutiny of IQC performance to accept assay runs with appropriate use of Westgard rules, modified where appropriate.
4
Regular review of IQC performance by monitoring coefficient of variation (CVs). This can also include cumulative CV analysis. Internal assessment of within batch and between batch CV. The expected CV will vary according to the technology used with, for example 3–5% for nephelometry or 8–12% for ELISA.
4
For flow cytometric assays one could expect even higher, depending on the antigen under investigation (for example we find CD4 + T-cells CV 6-10%; CD16 + NK cells CV 15-22% [RJL – unpublished data]). Appropriate root cause analysis investigations of IQC/EQA failure. The principle of optimising conjugated antisera for use in indirect immunofluorescence. See text for detail. In this example the 1 in 100 dilution shows prozone. The appropriate dilution for use is 1 in 400.
Overcoming clinical risk given measurement uncertainty with immunoassays
Given the known lack of standardisation, international calibrators or reference preparations available for immunoassays, it is clear that control procedures such as internal and external control and assessment should be scrutinised to minimise the potential risk.
‘Sense checking’ is very important to limit illogical results. This should be done at the assay authorisation stage. For example if a complete run of ANA or anti-TTG (anti-tissue transglutaminase) elicited no positive results this should flag as an oddity. Thus, if the expected positivity rate was approximately 10% for each (a typical figure, but which will vary depending on the population studied), one would statistically expect positive results in each run. For example, assuming 10% prevalence and a batch size of 100 samples, it is very unlikely only one would be positive (p=0.0007). It is important that results are looked at in combinations and not in isolation by suitably trained and qualified staff who have a sound analytical and clinical understanding of the assays. For example a dsDNA antibody should not be reported with a negative ANA without further analysis. This might include checking by alternate methods with higher specificity. Some tests require correlation with tests that may be performed in other disciplines, such as total IgA concentration with low IgA anti-TTG results or antimitochondrial antibody with liver function tests. This is good practice and, with the advent of combined blood sciences departments and integrated information technology solutions should become increasingly more common.
The use of clinical sensitivity and specificity gives good indications for the clinical significance of results for different clinical conditions. Where possible these should be publicised to users. However, many widely quoted clinical specificities particularly must be used with caution. The issue of technological drift must be considered. 25 Many of the clinical associations were defined using the original high specificity ‘gold standard’ assays. With the greatly improved sensitivity of modern methods there is often an adverse effect on the clinical specificity of autoantibodies for specific clinical conditions. For example high affinity dsDNA antibodies detected by the original radioimmunoassay (using the Farr technique) were highly specific for systemic lupus erythematosus but not sensitive. 26 Current highly sensitive ELISA assays to dsDNA also detect low affinity antibodies which may be found in other disease states and healthy, particularly elderly, individuals therefore reducing specificity. Sole reliance on data from manufacturers is not advised but large in-house studies to accurately determine clinical sensitivity and specificity may not be feasible. Furthermore, differences in local populations affect the pretest probabilities of having the disease under investigation and thus affect the determination of positive and negative predictive values. These locally derived parameters may be more useful in interpretation than the sensitivity and specificity alone.
It is not recommended that any Immunology test results alone are used to make a definitive diagnosis without correlation with the clinical context. Many parameters are prognostic indicators but again should not be used in isolation to direct clinical management. In the case of potentially useful serial markers (e.g. dsDNA antibodies, anti-glomerular basement membrane or ANCA titres) the use of reference change values (a statistical tool reviewed by Fraser 27 ) is likely to be very inaccurate given the assay uncertainty as well as the heterogeneity of the clinical conditions. However, delta checking for the antibody concentrations can be useful and enables suitable comments to be added to results. One clear example is the use of paraproteins as markers of response to therapy in plasma cell proliferative disorders. 28 Clinicians are advised to look at trends with results rather than absolute value differences.
Good professional practice requires us to define our uncertainty. This means knowing our assays, their limitations and not generating results blindly without putting them into context. Immunoassays may have a long way to go in standardisation but we can ensure we are providing results and interpretation to the best of our ability.
Footnotes
Acknowledgements
This article was prepared at the invitation of the Clinical Sciences Reviews Committee of the Association for Clinical Biochemistry and Laboratory Medicine.
Declaration of conflicting interests
None declared.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethical approval
Not applicable
Guarantor
RJL.
Contributorship
SCB and RJL both wrote, edited and agreed the final version.