
Abstract
Recent studies of inherited disorders of phosphate metabolism have shed new light on the understanding of phosphate metabolism. Phosphate has important functions in the body and several mechanisms have evolved to regulate phosphate balance including vitamin D, parathyroid hormone and phosphatonins such as fibroblast growth factor-23 (FGF23). Disorders of phosphate homeostasis leading to hypo- and hyperphosphataemia are common and have clinical and biochemical consequences. Notably, recent studies have linked hyperphosphataemia with an increased risk of cardiovascular disease. This review outlines the recent advances in the understanding of phosphate homeostasis and describes the causes, investigation and management of hypo- and hyperphosphataemia.
Introduction
Phosphorus in the form of inorganic or organic phosphate is a major component of all tissues and is essential for many functions within the body. It is a vital constituent of bone and cell membranes and of molecules such as adenosine triphosphate (ATP), nicotinamide adenine dinucleotide (NAD), cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP); it is essential for energy storage and metabolism and it is important for cell signalling and enzyme activation. Phosphate has been shown to be involved in osteopontin gene expression, chondrocyte apoptosis and vascular smooth muscle differentiation. 1
Phosphate homeostasis
The average healthy adult has approximately 25 mol (680 g) of phosphate; 85% is found in the skeleton, 10–15% in soft tissues and <1% in extra-cellular fluid (ECF). In healthy adults, phosphate balance is maintained by the kidneys and to some extent by the gastrointestinal tract. In children there is a positive balance of 2–3 mmol/day and in elderly subjects there is a small negative balance, primarily due to age-related bone loss.
Intestinal absorption of phosphate
Phosphate is present in high concentration in many foods especially dairy products, meat and vegetables. The average intake of phosphate is about 50 mmol/day, but can be higher in those consuming large quantities of meat or dairy products. Absorption of phosphate is greater when it is present in a soluble form such as in meat or dairy products than when it is in the insoluble form. About 7 mmol/day of phosphate is secreted in the gastrointestinal tract and some of this is reabsorbed. The total faecal excretion of phosphate is about 16 mmol/day giving a net absorption of 34 mmol/day, which is 60–70% of intake.
Phosphate absorption takes place throughout the small intestine and colon. Absorption from the colon has no physiological relevance, but is important in causing hyperphosphataemia during phosphate enemas. 2 There is no consensus on the major site of phosphate absorption in the small intestine as this depends on the study method used and the greatest absorptive capacity of phosphate in humans is in the duodenum. There is some evidence that the upper and lower segment of the intestine contribute equally to overall phosphate absorption.
It is generally accepted that there are two mechanisms of phosphate absorption: a sodium-dependent active pathway and a sodium-independent or non-active (passive) pathway. The contribution of the sodium-dependent pathway depends on the method used to study and the phosphate concentration in the lumen.
3
The non-active (passive) transport of phosphate occurs by paracellular transport via tight junctions between cells. The rate of absorption depends on the phosphate concentration of the lumen and is linearly related to it. The active transport occurs via the sodium-phosphate co-transporter NPT2b and possibly type III transporters PiT1 and PiT2.
4
NPT2b belongs to the SLC34 solute carrier family and is expressed in the brush border of enterocytes.5 It is electrogenic, has a stoichiometry of 3:1 for sodium and phosphate and has high affinity for phosphate. NPT2b appears to contribute to the majority of active transport.
6
PiT 1 and PiT 2 are type III co-transporters belonging to the SLC20 family of transporters. Although PiT1 and PiT2 are expressed in the intestinal cells, the contribution of these to phosphate absorption is minimal as sodium-dependent phosphate transport is virtually absent in NPT2b knock out mice.
3
The mechanism of active absorption of phosphate in the intestine is shown in Figure 1. Phosphate enters the enterocyte via NPT2b transporter utilizing the energy generated by the Na+/K+-ATPase on the basolateral membrane. The exact mechanism of phosphate efflux through the enterocyte into the ECF is not fully understood. It has been suggested that most of the active phosphate transport is mediated by NPT2b transporter and this pathway may only be important during fasting or with low dietary phosphate
7
. Under normal phosphate intake, sodium-independent (passive) transport is more important.
7
Transport of phosphate in the intestine. Phosphate is transported from the intestinal lumen via NPT2b. There is also paracellular transport between cellular tight junctions.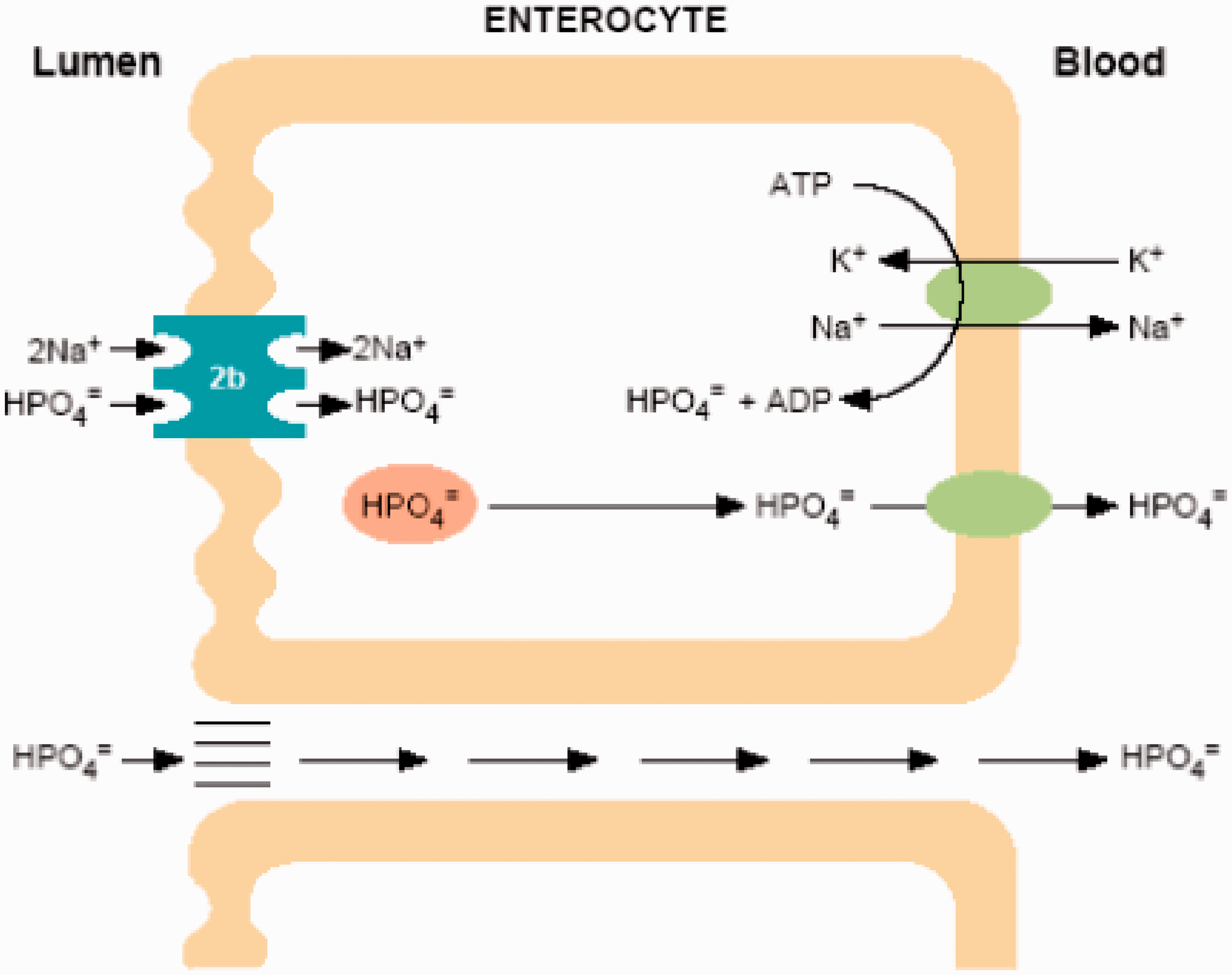
Regulation of intestinal transport
Dietary phosphate intake and 1, 25 dihydroxycholecalciferol (1, 25 DHCC) are important factors that regulate intestinal phosphate absorption. 8 Other factors include epidermal growth factor (EGF), glucocorticoids, oestrogens, metabolic acidosis and phosphatonins. 7 There is no evidence for a direct effect of PTH on phosphate transport in the gut. 9 Low dietary phosphate and 1, 25 DHCC stimulate phosphate absorption via the NPT2b transporter by different mechanisms. 1, 25 DHCC acts by inducing the transcription of NPT2b 10 while low phosphate diet acts via post-translational mechanisms. 11 The phosphatonin, fibroblast growth factor 23 (FGF23) influences intestinal absorption via its effects on the production of 1, 25 DHCC (further details below). 12 Matrix extracellular phosphoglycoprotein (MEPE), another phosphatonin, has a direct effect on intestinal phosphate transport, independently to changes in 1,25DHCC. 13 As MEPE is expressed in the intestinal tract it has been suggested that MEPE may act as an autocrine or paracrine factor influencing phosphate transport across the intestine.
Renal excretion of phosphate
About 90% of serum phosphate is ultrafilterable. Most of it is reabsorbed leaving 20–30 mmol/day to be excreted. Proximal convoluted tubule is the major site of phosphate reabsorption, accounting for 70% of the filtered load. Phosphate transport in the renal tubules is mediated via the type II sodium-phosphate co-transporters, NPT2a and NPT2c and to a much lesser extent by type III transporters. The highest concentration of NPT2a is expressed in the brush border membrane of the S1 segment of proximal convoluted tubule. 14 This electrogenic transporter has a high affinity for phosphate and preferentially transports it; NPT2c is electroneutral and has lower affinity for phosphate.
Studies using NPT2a knockout mice have shown that this transporter is responsible for 70% of phosphate reabsorption. 15 Studies in patients with mutations in the NPT2a and NPT2c genes suggest that NPT2c plays a significant role in maintaining phosphate balance.16,17 In mice where both the NPT2a and NPT2c have been knocked out, there is residual phosphate reabsorption suggesting that type III transporters PiT1 and PiT2 may have some roles in phosphate balance. 18
The mechanism by which phosphate is transported from the filtrate to the extracellular fluid is shown in Figure 2. Phosphate is transported into the cell by NPT2a and NPT2c. The rate of this transport depends on the number of transporters available and on the sodium gradient across the luminal membrane. The Na+/K+-ATPase in the basolateral membrane generates this sodium gradient. Exit of phosphate from the cell via the basolateral membrane is poorly understood.
19
Reabsorption of phosphate in the proximal tubule. Phosphate is transported across the brush border by NPT2a and NPT2c transporters. Phosphate entering the tubular cell mixes with the intracellular pool of Pi and is transported across the basolateral membrane by a poorly understood mechanism. The Na+/K+-ATPase on the basolateral membrane (not shown in figure) pumps Na+ out of the cell thereby providing the driving force for the luminal entry of sodium.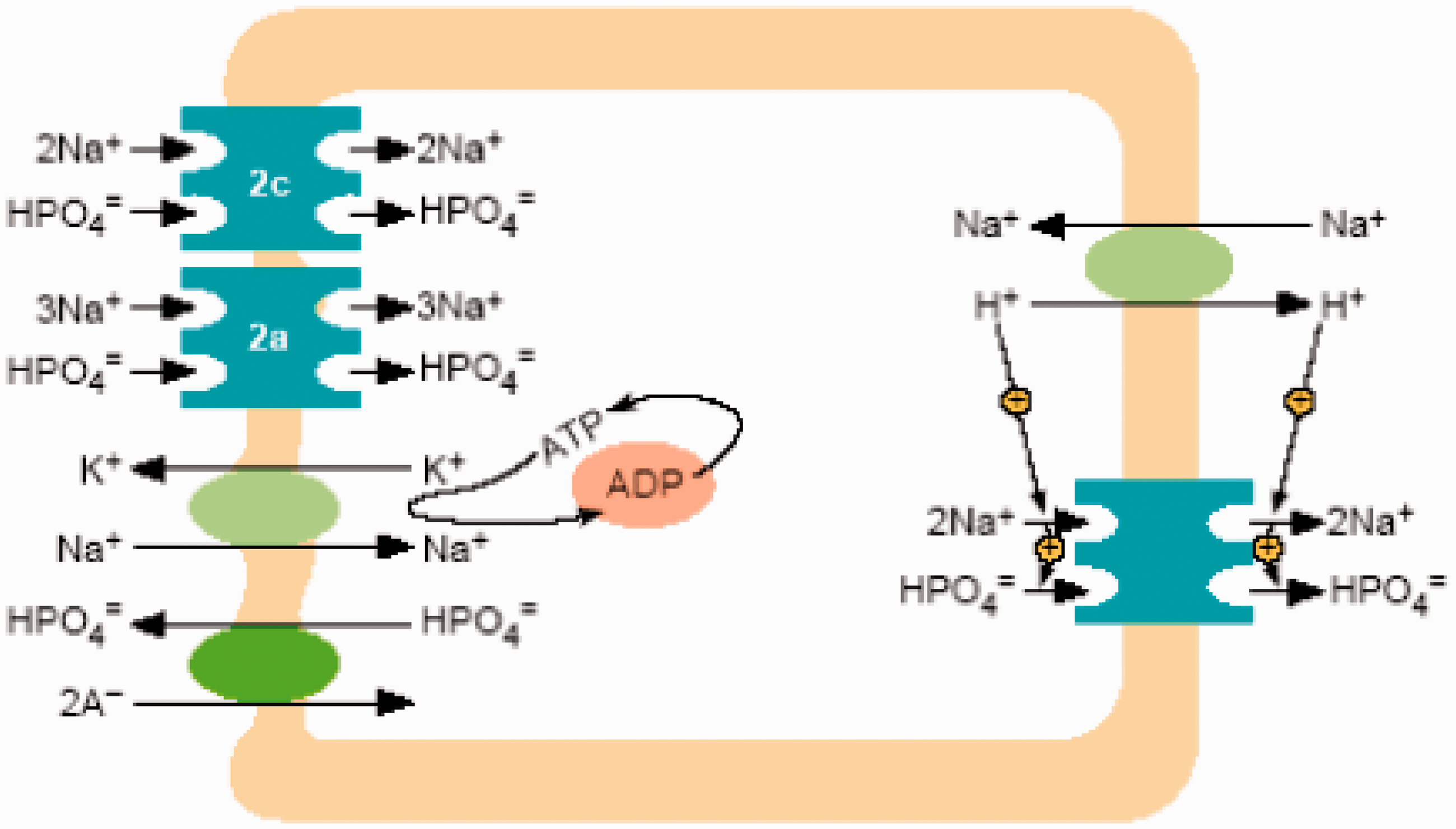
Regulation of renal phosphate reabsorption
Factors affecting phosphate reabsorption include dietary phosphate intake, hormones (principally PTH, 1,25DHCC and FGF23) and metabolic changes (acid-base disorders, ECF volume depletion).
Dietary intake of phosphate. Changes in dietary phosphate intake are followed by corresponding changes in renal excretion and this is brought about by changes in the expression of phosphate transporters (NPT2a, NPT2c and PiT2). Adaptive changes in the expression of NPT2a occur rapidly within 2 h without changes in plasma phosphate and are independent of PTH and 1, 25 DHCC.11,20 Changes in NPT2c and PiT2 take longer.
7
The presence of NPT2a in the brain suggests that the adaptive changes in phosphate reabsorption may be mediated via central transporters in the brain acting like phosphate sensors.
21
It has been shown that the Na+/H+-exchanger regulatory factor 1 (NHERF-1) is required for the renal adaptation by the NPT2a transporter
22
although the exact mechanism is not fully understood but may involve FGF23 (see below) and renal dopamine.
23
Parathyroid hormone. Parathyroid hormone (PTH) is a major regulator of phosphate reabsorption. PTH binds to PTHE1 receptors (also known as PTH/PTHrP receptor) which are highly expressed in renal tubules, and activates the adenylate cyclic AMP system as well as the phospholipase-C protein kinase (PKC) pathway. This results in rapid endocytosis and degradation of NPT2a transporters
24
and therefore appears to be an acute effect.
25
There is evidence that PTH also regulates NPT2c transporter.
26
1, 25 Dihydroxycholecalciferol. The role of 1, 25 DHCC on phosphate reabsorption and the interactions with PTH is not clear. 1, 25 DHCC appears to have a permissive role in the action of PTH on phosphate reabsorption. The effect of PTH on phosphate transport is blunted by vitamin D deficiency and restored by 1, 25 DHCC supplementation. There is some evidence that 1, 25 DHCC acts directly on the proximal tubule to stimulate phosphate reabsorption via the induction of NPT2a and NPT2c.
10
Phosphatonins. Phosphatonins are factors that regulate phosphate metabolism by reducing renal phosphate reabsorption. They include FGF23, secreted frizzled-related protein 4 (sFRP-4), fibroblast growth factor 7 (FGF7) and MEPE.
Fibroblast growth factor 23
FGF23 is a 30 kD glycoprotein released primarily by osteocytes and to a lesser extent by osteoblasts. 27 FGF23 shares a common core region with the FGF superfamily and is expressed on bone, thymus, lymph nodes and in the brain. 28 The FGF23 gene is located on chromosome 12p13. 29 This gene encodes a 251 amino acid protein that is cleaved and followed by O-linked glycosylation 30 by the enzyme UDP-N-acetyl-alpha-D-galactosamine:polypeptide N-acetylgalactosaminyl-transferase 3 (GALNT3).
Actions of FGF23
FGF23 reduces serum phosphate concentration by reducing renal phosphate reabsorption via reduction in the expression of the NPT2a transporter.31,32 In contrast to PTH, FGF23 inhibits 25 hydroxyvitamin D-1 hydroxylase and stimulates the 25 hydroxyvitamin D-24 hydroxylase instead. These effects occur earlier than changes in serum phosphate. 32 The effects of FGF23 on 1, 25 DHCC and serum phosphate were found to be independent of PTH. 32 Although parathyroid glands express FGF receptors and its co-factor Klotho, the effect of FGF23 on PTH secretion is not fully understood. It has been suggested that FGF23 and PTH may function as a feed back loop similar to the FGF23 and 1,25DHCC loop. However, this feed back loop may be modified by other local and systemic factors. 33
Regulation of FGF23
FGF23 is regulated by systemic (1, 25 DHCC and possibly phosphate) and local factors. 1, 25 DHCC regulates FGF23 by both vitamin D receptor (VDR) dependent and independent mechanisms. 34 FGF23 in turn suppresses 1, 25 DHCC. Thus, FGF23 and 1,25 DHCC form an endocrine negative feed back system. 33 Phosphate loading in mice has been shown to increase FGF23 concentration 35 and this effect is dependent on 1,25 DHCC and VDR. 33 In humans, the effect of oral phosphate on FGF23 is not clear as some studies have shown an increase in FGF23 following dietary loading 36 whereas others have shown a decrease. 37 The exact mechanism by which dietary phosphate influences FGF23 concentration is still not fully understood.
The role of PTH in the regulation of FGF23 remains controversial at present although FGF receptor and its cofactor Klotho are co-expressed in parathyroid glands. 38
Factors produced locally in the bone have a role in FGF23 regulation. Dentin matrix acidic phosphoprotein (DMP1), an inducer of mineralization of bone inhibits FGF23 by an unknown mechanism. 39 Phosphate regulating gene with homologies to endopeptidases on the X-chromosome (PHEX) belongs to the endothelin converting enzyme family and is highly expressed in osteocytes and osteoblasts. 33 Deletion of the PHEX gene leads to high FGF23, but the exact mechanism remains obscure. 33 MEPE has the opposite effect to DMP1. 40 Proteolytic cleavage of MEPE releases the C-terminal acidic serine-aspartate-rich MEPE (ASARM) peptide which has an inhibitory effect on mineralization and renal phosphate reabsorption. 34 MEPE and ASARM both bind specifically to PHEX in vitro and inhibit PHEX activity suggesting that they may influence FGF23 by decreasing PHEX activity. 41
Mechanism of FGF23 action
FGF23 binds to FGF receptors of which there are four (FGFR1-4). Alternative splicing of FGFR gene produces multiple FGFR isoforms. 42 FGFR1C is the major receptor involved in FGF23 action. 43 The action of FGF23 requires the co-expression of Klotho, a glucuronidase that acts as a cofactor. 44 Klotho is a type 1 membrane protein expressed in the distal convoluted tubules of the kidney, parathyroid gland and the choroid plexus. 45 Klotho binds to the c-splice isoform of FGF receptors. FGF23 binds to this FGF receptor: klotho complex 44 and activates the mitogen-activated protein kinase (MAPK) pathway. 46 The FGF receptor:klotho complex is predominantly seen in distal tubules 47 but the major action of FGF23 is on the proximal tubules which suggests an autocrine/paracrine pathway between the distal and proximal tubules. 48 FGFR1 and FGFR2 are expressed in osteoblasts which suggests a possible role for FGF23 in bone. 49
Other phosphatonins
The protein secreted frizzled protein 4 (sFRP4) is ubiquitously expressed and circulates as a 48 kD protein. As the expression of this gene is increased in tumour-associated osteomalacia, it has been suggested to be a phosphatonin. However, in sFRP-4 knock out mice, no significant effect on phosphate homeostasis was observed. 50 FGF7 has been shown to be increased in tumour-associated osteomalacia, but its exact role in phosphate homeostasis is yet to be determined. MEPE is a 525 amino acid protein expressed in bone, salivary gland and dental tissue and expressed in tumour-associated osteomalacia. 51
Recent evidence suggests that the intestinal mucosa may influence renal phosphate reabsorption via secretion of an as yet unknown factor. Infusion of phosphate solution into the duodenum causes a rapid increase in phosphate excretion by the kidney which is independent of PTH, FGF23, sFPR-4 or GFR. 52 This effect is not seen if phosphate is replaced with sodium chloride. It has been suggested that MEPE may be the ‘intestinal phosphatonin’. 6
Hormones and metabolic factors that regulate phosphate metabolism
Glucocorticoids in pharmacological doses down regulate phosphate transport. 53 Growth hormone and thyroxine increase phosphate transport. Insulin, calcitonin and EGF are also known to influence phosphate excretion. Intra-renal dopamine causes phosphaturia while serotonin reduces phosphate excretion. 54
Metabolic factors that influence phosphate metabolism include chronic metabolic acidosis, which causes a decrease in phosphate reabsorption 55 via an effect on NPT2a and NPT2c transporters. Respiratory alkalosis causes a reduction in phosphate excretion whereas respiratory acidosis has the opposite effect. Plasma calcium decreases phosphate excretion by a direct effect as well by an indirect effect via PTH. ECF volume expansion causes phosphaturia possibly via renal dopamine.
Plasma phosphate
The total plasma phosphate concentration is 3.9 mmol/L of which only 0.8–1.3 mmol/L is inorganic, the rest is present as organic compounds including phospholipids. Inorganic phosphate in serum exists in three forms: ionized (about 55%), complexed to cations (about 35%) and protein bound (about 10%). Approximately 90% of plasma inorganic phosphate is ultrafiltrable, i.e. non-protein bound fraction. 56 The relative proportion of HPO42− and H2PO4− depends on the pH, and at normal pH the ratio is 4:1. This ratio decreases in acidic conditions and increases in alkaline states.
Intracellular phosphate is mostly in the organic form – as intermediates of carbohydrates or as lipids and proteins. The concentration of inorganic phosphate in the cells is about 4–5 mmol/kg of tissue water, but most of this is bound, sequestrated in organelles or otherwise immobilized. Using NMR spectroscopy, the free inorganic phosphate concentration has been estimated to be about 1 mmol/L.
Plasma phosphate concentration shows a marked diurnal rhythm – lowest in the morning and highest at night. Fasting can abolish this diurnal variation. 57 Plasma phosphate concentration is age-dependent. It is high at birth and increases further during the first few hours of birth, and then declines after the first day of life. Children have higher phosphate concentration than adults. There is no gender difference in phosphate concentration in the age group 20–40 years. Phosphate concentration is lower in older men than in women. 58 Plasma phosphate decreases during pregnancy. 59
Measurement of plasma and urine phosphate
Phosphate is commonly measured by methods based on the reaction of phosphate ions with ammonium molybdate to form a phosphomolybdate complex – (NH4)3 P(Mo3O10)4 which can be measured directly at 340 nm56 or after reduction to a complex heteropolymer, molybdenum blue that is measured at 600–700 nm. Reducing agents used include aminonaphtholsulphonic acid, ferrous ammonium sulphate, stannous chloride, ascorbic acid and semidine hydrochloride.
Other uncommonly used methods include the vanadate–molybdate and enzymatic methods. 56 The vanadate–molybdate method, which is carried out at an acidic pH, tends to give higher results due to the hydrolysis of organic phosphate esters. Enzymatic methods are available but not in widespread routine use.
There is no reference method for the measurement of serum phosphate at present. The American Association for Clinical Chemistry (AACC) has proposed a ‘selected method’ based on ammonium molybdate with semidine hydrochloride as the reducing agent. However, the direct molybdate method measured at 340 nm (UV) is more commonly used in many discrete analysers because of its speed, good precision, accuracy and reagent stability. To prevent protein precipitation, a solubilizing agent such as Tween-80 is added to the reagents. Sample blanking is necessary to correct interference from other compounds, which absorb at 340 nm. Bichromatic analysis with 340 and 380 nm analysis is helpful to reduce interference. 56
Regarding the analytical goal, based on the biological variability, the required analytical precision for serum phosphate for medical needs is between 2.9 and 5.0%. 60 According to UKNEQAS data, most of the current methods are capable of achieving this goal.
Hypophosphataemia
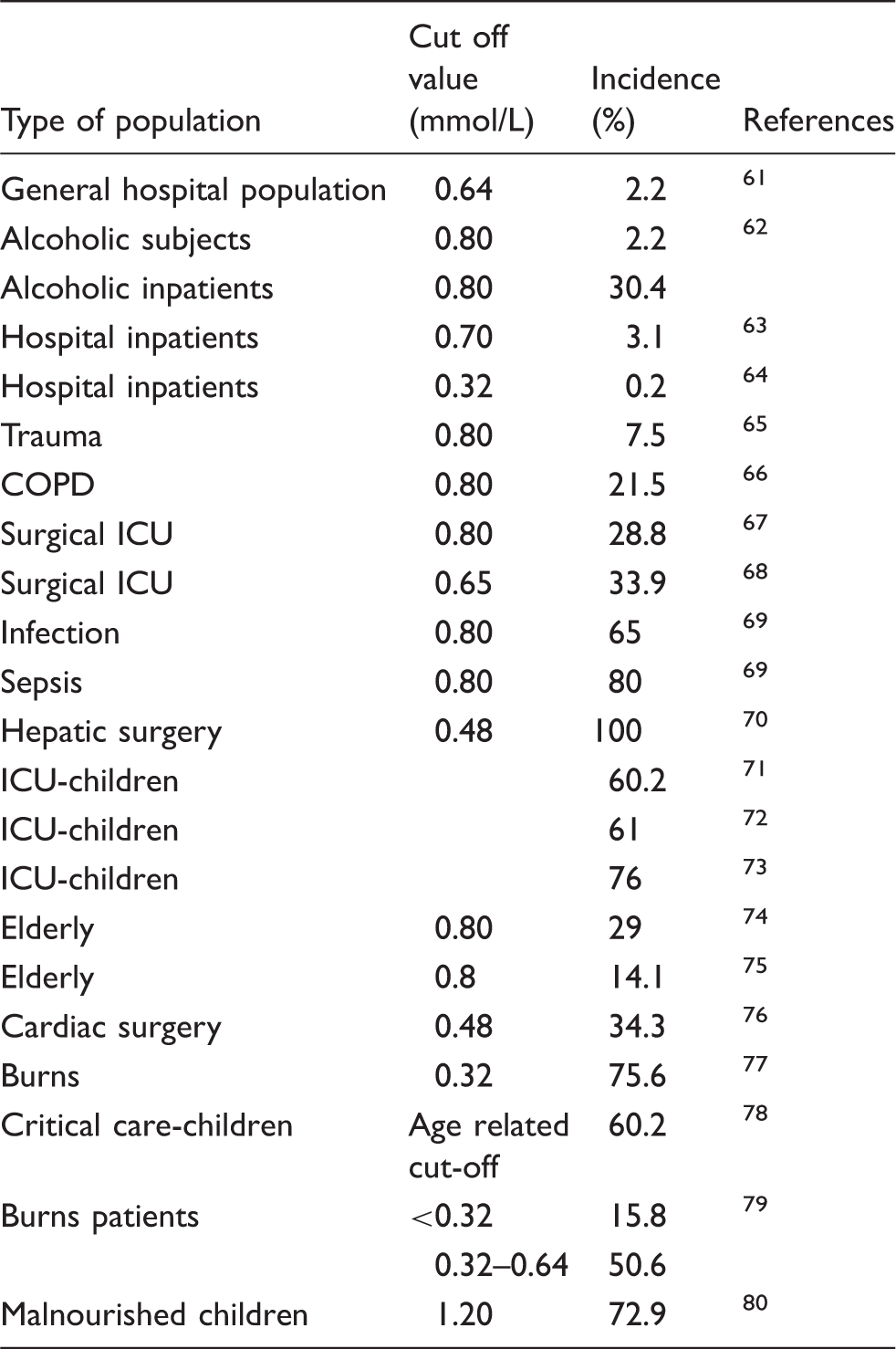
Incidence of hypophosphataemia in different patient populations.
Causes of hypophosphataemia
Causes of hypophosphataemia.
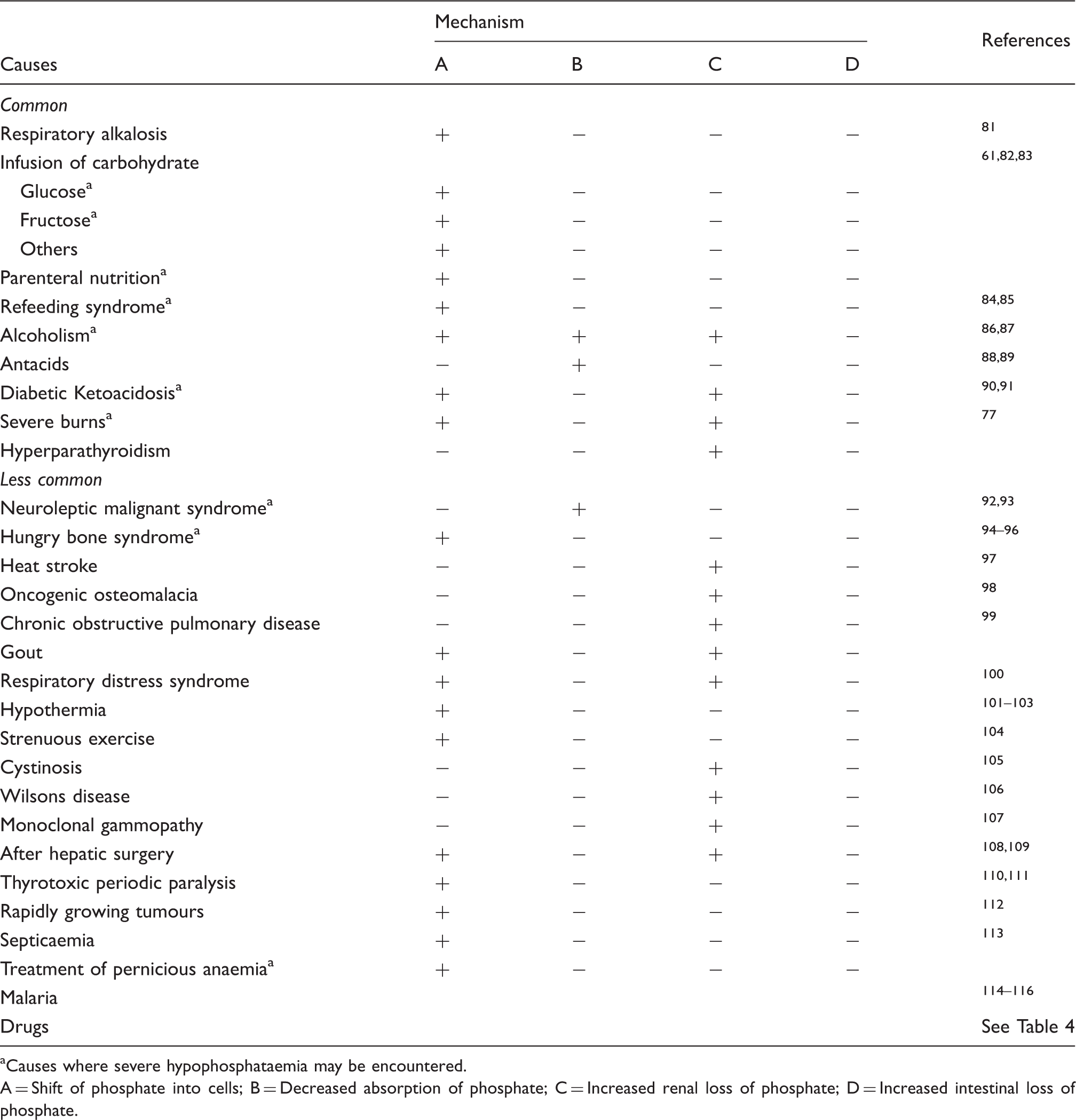
Causes where severe hypophosphataemia may be encountered.
A = Shift of phosphate into cells; B = Decreased absorption of phosphate; C = Increased renal loss of phosphate; D = Increased intestinal loss of phosphate.
Pseudohypophosphataemia
Spuriously low serum phosphate can arise due to analytical or pre-analytical factors. Gross elevation of leucocytes can cause hypophosphataemia in addition to hypokalaemia. 148 High bilirubin concentration has been shown to interfere in the bichromatic analysis of phosphate 149 but this was not seen in methods where a reducing agent was used. 150 Mannitol when given in large doses can bind with molybdate and cause pseudohypophosphataemia especially when the concentration of molybdate is low. 151
Reduced absorption
It is unusual for reduced dietary intake alone to cause hypophosphataemia because there is usually renal and/or intestinal adaptation. Malabsorption or phosphate binding drugs can cause hypophosphataemia. 152 It takes approximately three months for hypophosphataemia to develop in healthy subjects given a low phosphate diet and antacids. 153 Long-term use of antacids can lead to osteomalacia. 154
Transcellular shift
Respiratory alkalosis and administration of carbohydrates, which are the commonest causes of hypophosphataemia, result from shift of phosphate into cells. Alkalosis, both respiratory and metabolic, causes a decrease in serum phosphate; however, the effect is greater with respiratory alkalosis. 155 Respiratory alkalosis causes intracellular CO2 to decrease, causing an intracellular alkalosis, which increases glycolysis, via stimulation of the key glycolytic enzyme phosphofructokinase. This leads to a reduction in intracellular phosphate and a consequent shift of phosphate into cells. In respiratory alkalosis serum phosphate can fall to <0.30 mmol/L. 155 Serum phosphate falls within 20 min of hyperventilation and it persists for 90 min after the ventilation returns to normal. 156 Hypophosphataemia associated with septicaemia, severe liver disease, heat stroke, salicylate intoxication, acute gout and malignant neuroleptic syndrome are all most likely to be due to the associated hyperventilation.
Carbohydrate administration has been shown to account for 40–43% of cases of hypophosphataemia.61,83 Even small amounts of intravenous glucose (4% or 5% dextrose) can cause significant hypophosphataemia. 157 If there is accompanying hyperventilation, the fall in serum phosphate is greater. 156 The degree of hypophosphataemia is related to the amount of carbohydrates infused. 158 Infusion of carbohydrates increase insulin release, which causes a shift of phosphate into cells.
Hypophosphataemia is exaggerated in patients who have been starving before the administration of carbohydrates, the so-called refeeding syndrome. This syndrome was described in malnourished prisoners of the Second World War and may have contributed to death in this group. Refeeding syndrome is seen in malnourished patients, after major surgery and in those with anorexia nervosa and may develop with oral, enteral or parenteral feeding. 82 Hypokalaemia, hypomagnesaemia as well as hypophosphataemia are all features of this syndrome. Decrease in serum phosphate is due to rapid uptake of phosphate into cells as a result of increased intracellular metabolism and the stimulation of phosphate uptake by insulin. 159
Serum phosphate decreases during treatment of diabetic ketoacidosis (DKA) 90 because of reduced intake of phosphate and increased urinary loss. At presentation, serum phosphate is normal or elevated as a result of release from cells due to increased catabolism. With rehydration and insulin therapy, plasma phosphate concentration rapidly decreases. 90
Increased cellular uptake of phosphate is responsible for the hypophosphataemia seen in patients with leukaemia, in those with rapidly dividing tumour cells after blood stem cell transplantation, and after parathyroidectomy. The latter is usually referred to as the ‘hungry bone syndrome’. 95
Hypophospataemia is also common in acute and chronic alcoholism due to a combination of factors: reduced intake (poor diet, anorexia, gastritis, vomiting and diarrhoea), increased loss (induced by alcohol, acidosis and associated magnesium depletion) as well increased cellular uptake (hyperventilation, intravenous glucose and bicarbonate, catecholamine release and anabolic state during recovery). 86
Increased loss
Hypophosphataemia after renal transplantation
Factors contributing to the hypophosphataemia after renal transplantation.

Primary, secondary and tertiary hyperparathyroidism cause an increased urinary loss of phosphate leading to hypophosphataemia. Hypophosphataemia in vitamin D deficiency is usually more severe than that in primary hyperparathyroidism because of the relatively higher PTH concentration in the former.
Decreased reabsorption of phosphate by proximal tubules is seen in renal tubular disorders such as Fanconi syndrome. Fanconi syndrome is caused by congenital diseases such as cystinosis and Wilson disease, monoclonal gammopathy, metabolic disorders, heavy metal poisoning, drugs such as tenofovir and aristocholic acid (found in Chinese herbal medicines) 166 and toluene poisoning. 167
Hypophosphataemia is seen in about 25% of patients with chronic obstructive pulmonary disease (COPD) probably due to the effects of prescribed drugs. 99
Hypophosphataemia is also common in patients with extensive burns 79 and is due to loss of phosphate by extravasation of serum, respiratory alkalosis and increased secretion of catecholamines. 168
Hypophosphataemia after hepatic surgery
Hypophosphataemia is observed almost invariably after hepatic surgery. 108 Serum phosphate starts to decrease on the first or second post-operative day and returns to normal by the ninth day. 169 The mechanism for hypophosphataemia post-hepatic surgery is not known, but it has been suggested that uptake of phosphate by rapidly regenerating liver cells may be a possible cause. This is supported by the correlation between hypophosphataemia and liver regeneration 170 although another study suggests that the quantity of phosphate taken up by regenerating liver cannot account for the degree of hypophosphataemia. 109 Renal loss has also been suggested as a possible mechanism although it has been demonstrated that this is independent of FGF23, FGF7, sFRP4 or PTH-related effects.169,171 The significance of hypophosphataemia after hepatic surgery has been debated. Some studies suggest that low phosphate indicates better prognosis, 172 while others suggest that it may delay postoperative recovery. 70 However, replacement of phosphate has been shown to improve recovery of hepatic function. 173
Tumour induced osteomalacia or oncogenic osteomalacia
This is a paraneoplastic syndrome due to secretion of FGF23 by some tumours. Features include muscle and bone pain, muscle weakness, osteomalacia (or rickets) associated with renal phosphate wasting, hypophosphataemia and inappropriate 1, 25 DHCC concentrations with normal serum calcium and serum 25OHD. Removal of the tumour leads to reversal of these features. In some cases the tumour is occult and difficult to locate or the tumour may take several years to manifest clinically. Functional imaging, for example using F18-fluorodeoxyglucose positron emission tomography or octreotide scintigraphy may help to locate the tumour. Tumours causing tumour-induced osteomalacia (TIO) include mesenchymal tumours, prostate cancer, oat cell carcinoma, haematological malignancies, neurofibromatosis, epidermal nevus syndrome and polyostic fibrous dysplasia of bone. 174
Drug-induced hypophosphataemia
Drugs causing hypophosphataemia.
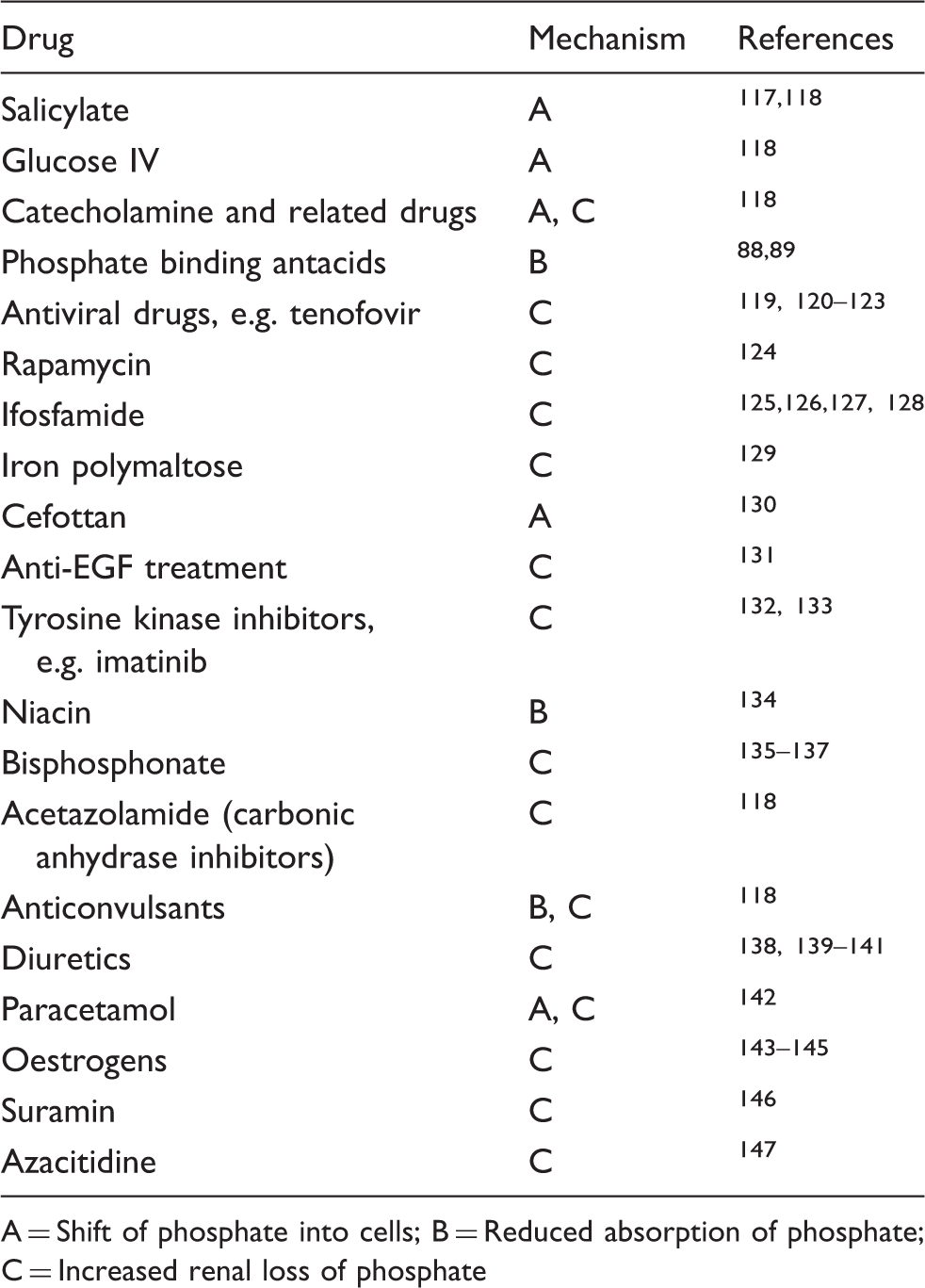
A = Shift of phosphate into cells; B = Reduced absorption of phosphate; C = Increased renal loss of phosphate
Genetic disorders of phosphate homeostasis.
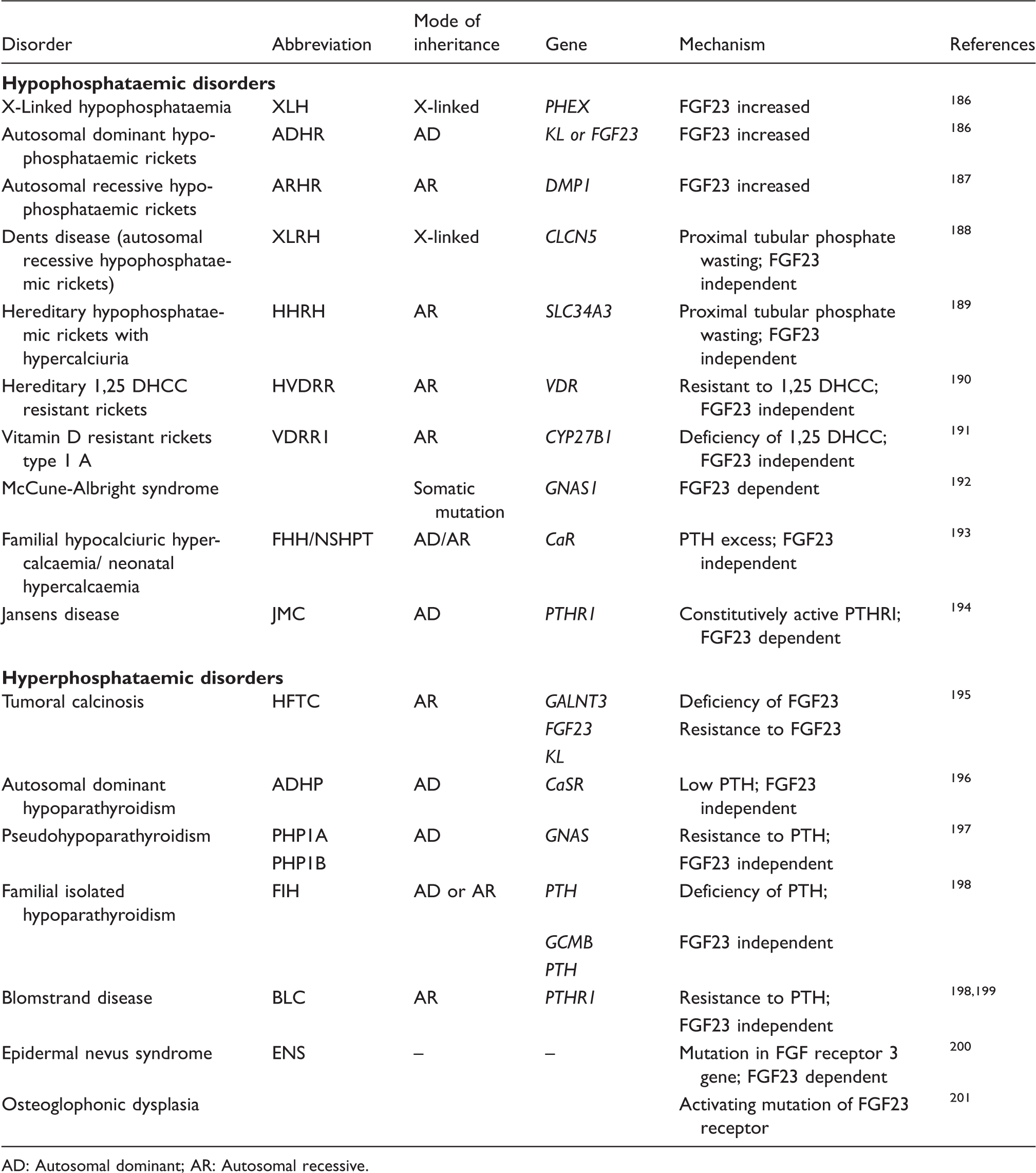
AD: Autosomal dominant; AR: Autosomal recessive.
Consequences of hypophosphataemia.

Genetic disorders causing hypophosphataemia
The inherited causes of hypophosphataemia are listed in Table 5 and briefly described below.
Laboratory findings in selected hypophosphataemic disorders.
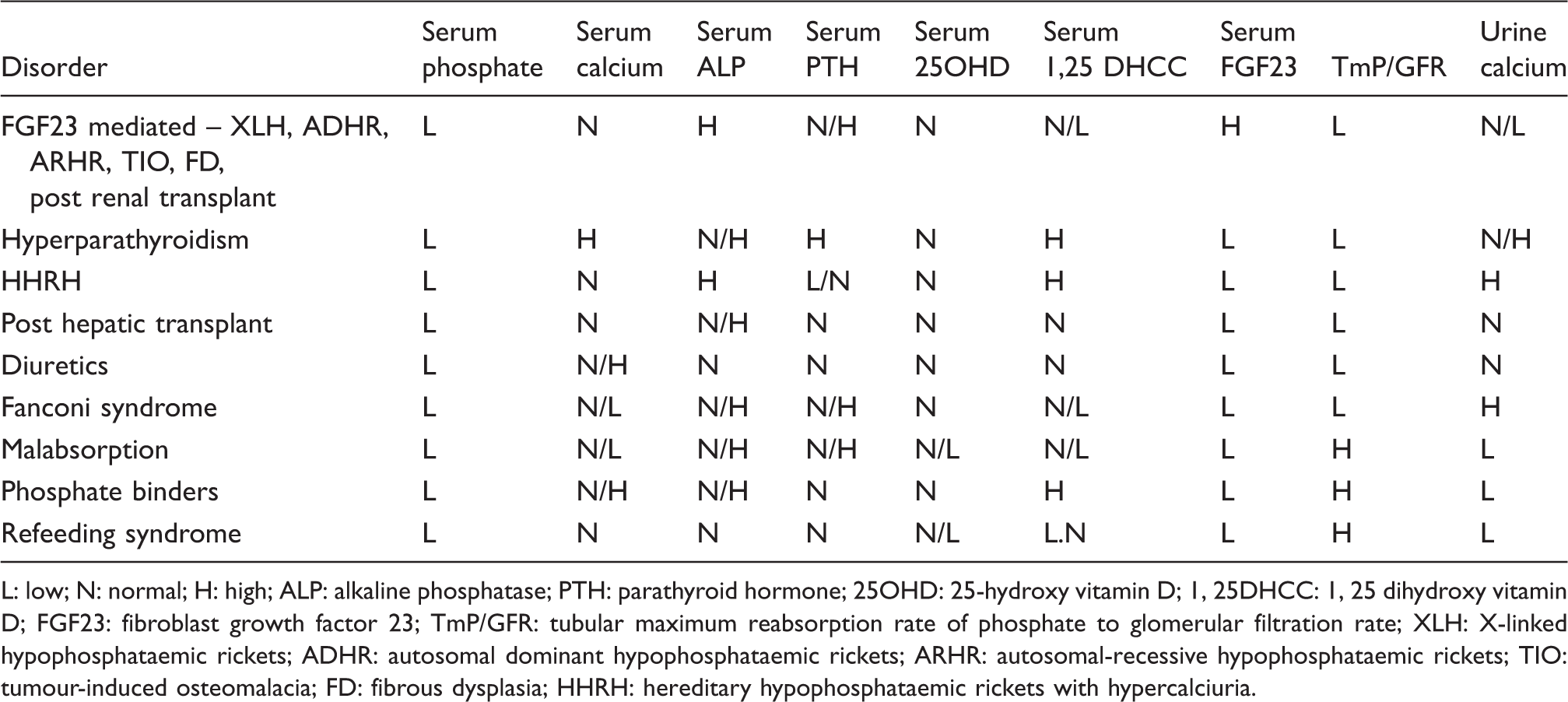
L: low; N: normal; H: high; ALP: alkaline phosphatase; PTH: parathyroid hormone; 25OHD: 25-hydroxy vitamin D; 1, 25DHCC: 1, 25 dihydroxy vitamin D; FGF23: fibroblast growth factor 23; TmP/GFR: tubular maximum reabsorption rate of phosphate to glomerular filtration rate; XLH: X-linked hypophosphataemic rickets; ADHR: autosomal dominant hypophosphataemic rickets; ARHR: autosomal-recessive hypophosphataemic rickets; TIO: tumour-induced osteomalacia; FD: fibrous dysplasia; HHRH: hereditary hypophosphataemic rickets with hypercalciuria.
Autosomal dominant hypophosphataemic rickets (ADHR) is an autosomal dominant disorder with features similar to XLH (see Table 7). This condition is due to a missense mutation in the FGF23 gene, which leads to alteration in the RXXR furin protease recognition site thereby rendering FGF23 resistant to proteolytic cleavage. 186 The expression of the disease can vary with children presenting with features similar to XLH, whereas adults may present with features of oncogenic osteomalacia. 187 Treatment is similar to XLH.
Autosomal-recessive hypophosphataemic rickets (ARHR) is a recently described disorder seen in children of consanguineous parents. The clinical presentation is similar to XLH with the exception of a relatively higher bone mineral density (BMD). 186 ARHR is due to a mutation in the gene encoding the protein DMP1, which is thought to suppress FGF23 secretion. 187
Dent’s disease (X-linked recessive hypophosphataemic rickets, XLRH) is a X-linked disorder due to a mutation in the voltage-gated chloride channel gene. 188 It is characterized by proximal tubular dysfunction leading to low molecular weight proteinuria, hypercalciuria and hypophosphataemia. These patients develop rickets or osteomalacia and nephrolithiasis or nephrocalcinosis.
Hereditary hypophosphataemic rickets with hypercalciuria (HHRH) is a rare autosomal recessive disorder characterized by hypophosphataemia, rickets, hypercalciuria and renal phosphate wasting. It is due to a mutation in the gene encoding the NPT2c transporter. FGF23 is not raised and the hypophosphataemia causes the expected increase in 1, 25 DHCC which in turn causes increased intestinal calcium absorption and hypercalciuria. 189
Hereditary 1, 25 DHCC resistant rickets is a rare disease caused by mutation in the VDR gene. It is also referred to as vitamin D-dependent rickets type 1, pseudo vitamin D-resistant rickets type II, calcitriol resistant rickets or hypocalcaemic vitamin D-resistant rickets. 190 Mutation in the VDR gene leads to partial or complete resistance to 1,25 DHCC leading to hypocalcaemia, secondary hyperparathyroidism and hypophosphataemia.
Vitamin D-resistant rickets type 1 A is a rare autosomal recessive condition due to a mutation in the gene for 1-alpha hydroxylase enzyme leading to 1, 25 DHCC deficiency. It is also called pseudovitamin D deficiency. The patients with this disorder are hypocalcaemic, hypophosphataemic and have rickets. 191
Fibrous dysplasia and McCune-Albright syndrome is a disease characterized by fibrous skeletal lesions and bone mineralization defects. Some patients develop skin pigmentation and premature sexual development. The latter constellation of symptoms is also described as McCune-Albright syndrome. Some patients with this syndrome develop hypophosphataemia, have a raised FGF23 concentration and a decreased tubular maximum for phosphate corrected for GFR (TmP/GFR), which is indicative of renal phosphate wasting. This disorder is due to an activating mutation in the signalling protein GS alpha. 192
Consequences of hypophosphataemia
The consequences of hypophosphataemia are listed in Table 6.
Acute effects
Hypophosphataemia has been reported to cause a number of acute effects especially when it is severe and prolonged. However, mild to moderate hypophosphataemia seldom causes any clinical effects. Most of these acute effects were described in the 1970 s in patients given total parenteral nutrition without adequate phosphate supplementation. 202
Biochemical effects
The acute effects are due to intracellular depletion of phosphate which in turn leads to a decrease in metabolites such as ATP and 2,3DPG. This causes generation of ATP from ADP and a consequent increase in AMP. The AMP generated is then converted to adenosine by 5′nucleosidase resulting in increased oxypurine production 109 and increased excretion of nucleotide metabolites. Production of 2,3DPG in red cells is reduced as direct consequence of a decrease in cellular phosphate. 158
Clinical effects
In experimental studies, hypophosphataemia was shown to cause rhabdomyolysis. 203 Hypophosphataemia-induced rhabdomyolyis is usually reported in patients with alcoholism, anorexia nervosa, theophylline toxicity, acute barium poisoning, toluene sniffing and after gastric surgery.204,205 In one study, increased CK was reported in 36% of hypophosphataemic patients. 206 ATP is essential for the maintenance of red cell shape, deformability and normal life span, and depletion of ATP therefore leads to destruction of red cells, anaemia and reticulocytosis.207,208 The decrease in 2,3DPG is directly related to the magnitude and duration of hypophosphataemia 83 and potentially reduces the delivery of oxygen to cells. Reduction in white cell ATP has been shown in vitro and in experimental studies to cause abnormalities in leucocyte function.202,209 Reduction in platelet ATP can cause thrombocytopenia, bleeding tendency and poor clot retraction. 202
Hypophosphataemia has been reported to cause respiratory failure and respiratory muscle weakness 210 that improved with phosphate supplementation. 211 In ventilated patients, hypophosphataemia caused a delay in weaning from ventilation. 212
Reduction in myocardial contractility has been demonstrated in experimental studies 213 and in inpatients 214 but only when serum phosphate was lower than 0.3 mmol/L. 215
Neurological abnormalities such as irritability, paraesthesia, confusion, convulsion and coma have been reported in severe hypophosphataemia associated with total parenteral nutrition. 216 Hypophosphataemia is associated with decreased insulin sensitivity. 217
Chronic effects
Chronic hypophosphataemia can cause osteomalacia or rickets. 178 Mortality in patients with hypophosphataemia has been reported to be nearly four-fold higher. 87 In the elderly, even a moderate degree of hypophosphataemia is associated with increased mortality. 74 However, these studies have been criticized for not taking into account confounding variables such as demographic factors, comorbidity and nutritional status. 218 In a recent study, hypophosphataemia was shown to be a marker of severity of illness and not as an independent predictor. 219
Investigation of hypophosphataemia
History and physical examination will help to exclude several causes. To determine if renal loss is a factor causing hypophosphataemia, indices of phosphate excretion such as tubular maximum reabsorption rate of phosphate to glomerular filtration rate (TmP/GFR) is required. The phosphate excretion index or index of phosphate excretion is no longer in use. 220
The ratio of the maximum rate of tubular phosphate reabsorption to the glomerular filtration rate (TmP/GFR) is considered the most convenient way to evaluate renal phosphate transport.
220
It corresponds to the theoretical lower limit of plasma phosphate below which all filtered phosphate would be reabsorbed. Although direct measurements of PTH, which increases renal phosphate excretion have replaced much of the utility of TmP/GFR measurements, it may still be useful in assessing renal reabsorption of phosphorus in a variety of pathological conditions associated with hypophosphatemia.
Nomogram to determine tubular maximum reabsorption rate of phosphate to glomerular filtration rate (TmP/GFR) given plasma phosphate (PO4) concentration and tubular reabsorption of phosphate (TRP). On the vertical axes, the inner scale is in mmol/L, whereas the outer scale is in mg/100 mL. TRP is calculated as detailed in text. A straight line joining plasma phosphate concentration, TRP and the right vertical axis gives the TmP/GFR.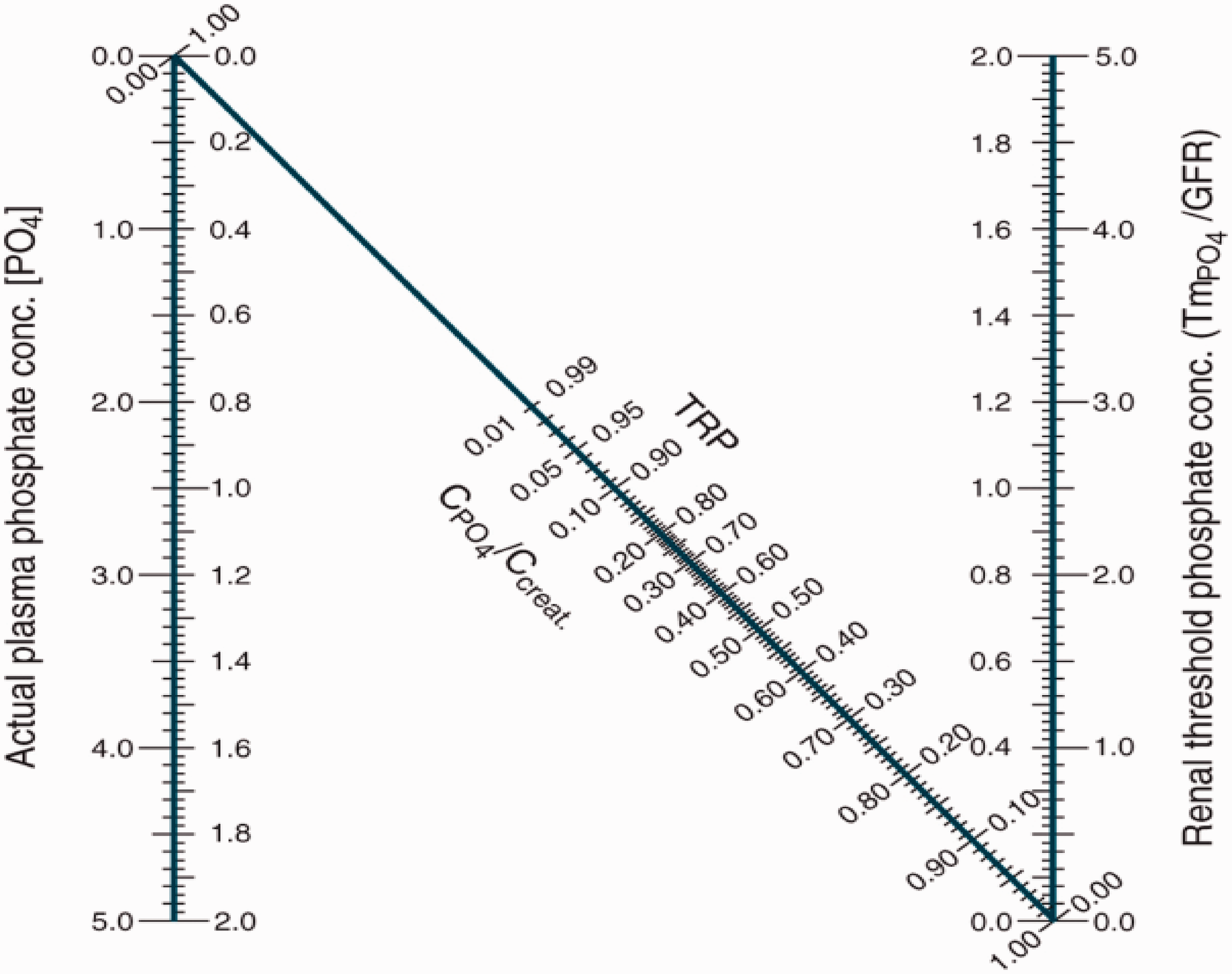
The tubular reabsorption of phosphate (TRP) is the fraction (or percent) of filtered phosphate that is reabsorbed by renal tubules. In general, a reduced TRP in the presence of hypophosphatemia is indicative of a renal defect in phosphate reabsorption. The fractional TRP can be used to calculate TmP/GFR as follows:
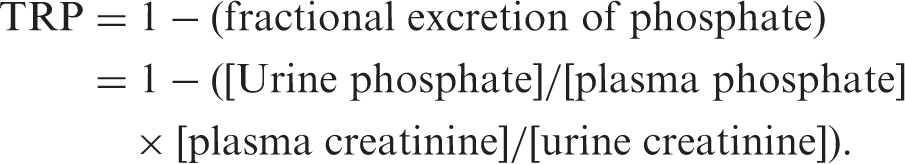


The measurement of serum PTH, 25OHD and sometimes serum 1, 25 DHCC may be necessary. A low serum 1, 25 DHCC in the presence of adequate 25OHD should alert to the possibility of a FGF23-mediated hypophosphataemia. The measurement of FGF23 is helpful in such situations but the test is not available widely. Urine amino acids, protein and glucose excretion indicate a proximal tubular disorder such as Fanconi syndrome. 221
Table 7 shows some laboratory investigations and their interpretation in phosphate disorders.
Management of hypophosphataemia
In situations where hypophosphataemia is likely, preventive measures should be taken. For patients on parenteral nutrition, adequate phosphate supplementation should be provided. If the hypophosphataemia is mild or moderate and of short duration, e.g. during treatment of DKA, no treatment is usually necessary especially if the patient is able to eat. In many cases, treatment of the underlying cause is sufficient. If the hypophosphataemia is severe or is of long duration, treatment with phosphate supplements should be considered. In patients with moderate hypophosphataemia, oral supplements may be adequate. Skimmed milk, which has 30 mmol/L of phosphate, is a good way of giving phosphate as it avoids diarrhoea seen with oral phosphate medication. Effervescent phosphate containing 16 mmol of phosphate per tablet (Phosphate-Sandoz) is available commercially.
Treatment regimes for hypophosphataemia.
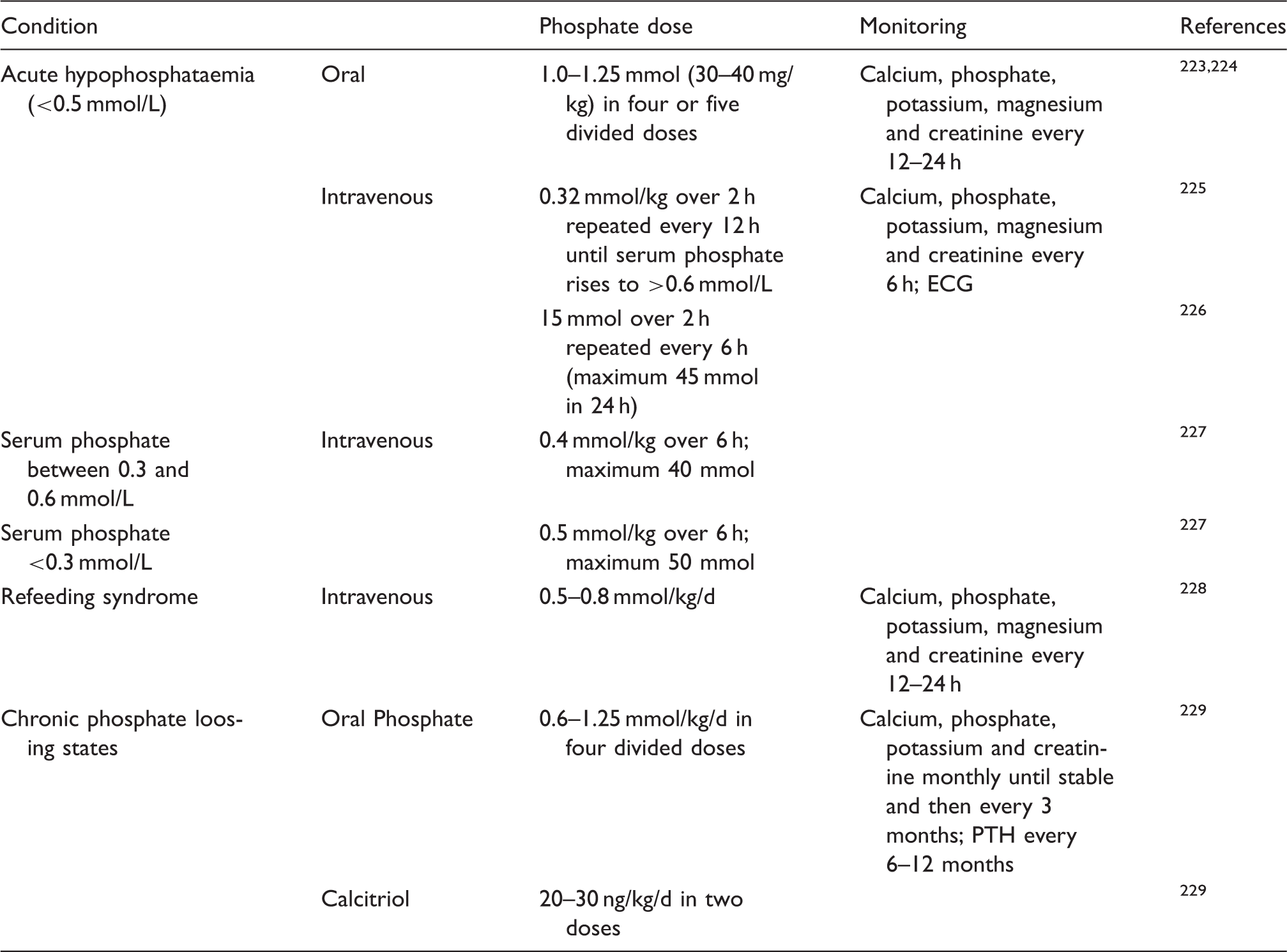
Complications of phosphate therapy include diarrhoea especially if the starting dose is high; hypocalcaemia due to precipitation of calcium in soft tissues; hyperkalaemia because most of the phosphate preparations contain potassium; and fluid overload. 221 To reduce the risk of diarrhoea, the dose should be increased gradually over 1–2 weeks. 221
Hyperphosphataemia
The incidence of hyperphosphataemia in hospitalized patients is estimated to be 15%; however, if renal disorders are excluded, the incidence is about 1.75% in inpatients and 1.5% in outpatients. 61
Causes of hyperphosphataemia
Causes of hyperphosphataemia.

Pseudohyperphosphatemia
Haemolysis, jaundice and lipaemia can interfere in some methods of phosphate measurement and cause pseudohyperphosphatemia. 231 If blood samples are left to stand for prolonged periods (usually more than 4–6 h), phosphate will move out of blood cells and cause a false elevation. 231 Pseudohyperphosphataemia has been described in patients given liposomal amphotericin B. 232 Possible mechanisms include interference in light scattering as result of a breakdown product of the liposome, release of phosphate from organic phosphate in liposome phospholipids and interference with the kinetic profile of the mannitol containing reagent system in some methods. 233 In chronic kidney disease (CKD) patients on haemodialysis, heparin can cause pseudohyperphosphatemia. 234
Paraproteinaemia is a well-recognized cause of pseudohyperphosphatemia due to the phosphate-binding effect of positively charged paraproteins.235,236 In patients with paraproteinaemia, purine nucleoside phosphorylase-based enzymatic methods or dry chemistry-based methods (where protein is removed before analysis) will give true results compared to chromogenic methods. 231 Recently, a case of pseudohyperphosphatemia in a haemodialysis patient due to contamination of saline used for dilution of sample was reported. 237
Increased phosphate intake
A very high intake of phosphate can lead to hyperphosphataemia especially if the renal function is poor. Ingestion of phosphate containing laxatives can lead to hyperphosphataemia and fatalities have been described. 238 Absorption of phosphate from phosphate-containing enemas can lead to severe hyperphosphataemia and even death. 239 Hyperphosphataemia and hypocalcaemia may develop in infants fed undiluted cow’s milk which has a high phosphate concentration. Vitamin D intoxication can cause hyperphosphataemia due to increased absorption of phosphate from the gastrointestinal tract and reduced renal phosphate excretion as a result of suppression of PTH caused by hypercalcaemia.
Transcellular shift
Tumour lysis syndrome due to lysis of tumour cells by cytotoxic drugs can cause severe hyperphosphataemia. 240 This syndrome is typically seen during treatment of acute lymphoblastic leukaemia, probably due to the higher phosphate content in blast cells. 241 A similar syndrome has been described after anti-infective treatment of leishmaniasis. 242 Rhabdomyolysis due to trauma, drug, toxins, hypoxia and infection can cause the release of phosphate from muscle cells. 243 Malignant hyperpyrexia following exposure to anaesthetic agents in susceptible individuals has also been reported to cause hyperphosphataemia. 244 Massive haemolysis can also cause a similar syndrome. 245
Shift of phosphate out of cells into the extracellular fluid (ECF) causing hyperphosphataemia is seen in untreated DKA 90 and lactic acidosis. Hyperphosphataemia tends to be more severe in lactic acidosis probably due to the effect of tissue hypoxia causing breakdown of ATP. 246
Reduced renal excretion
Renal failure is the commonest cause of hyperphosphataemia and is seen in more than 70% of patients with CKD. In patients with CKD, the capacity to excrete phosphate decreases due to the reduction in the number of functioning nephrons. 247 In the early stages of CKD, serum phosphate concentration is maintained due to an increase in PTH and FGF23. 248 Serum FGF23 rises before PTH or serum phosphate, 249 but the mechanism of increase in FGF3 in CKD is not fully understood. As the GFR falls, these adaptive mechanisms are inadequate to maintain phosphate balance and serum phosphate starts to rise when the GFR falls below 30 mL/min. 250 High concentration of FGF-23 leads to reduced 1, 25 DHCC and this contributes indirectly to secondary hyperparathyroidism. Recently, it was shown that serum FGF23 is a predictor of refractory secondary hyperparathyroidism in end stage renal disease 251 and a marker of the efficacy of calcitriol treatment. 252 The high concentration of FGF23 may contribute to the bone demineralization in CKD patients. 253 High FGF23 in CKD is associated with increased mortality independent of other factors. 254
Decreased renal excretion may be due to reduced PTH as in hypoparathyroidism which may be due to surgical removal of the parathyroid glands, autoimmune destruction of parathyroid glands or due to inherited disorders.
Genetic causes of hyperphosphataemia
There are several genetic causes of hyperphosphataemia as listed in Table 5 and discussed below.
Familial tumoral calcinosis (FTC) is a rare autosomal recessive disorder characterized by ectopic calcification in the major joints and vasculature. FTC is due to a mutation in the FGF23 gene, GALNT3 gene (FGF23 is glycosylated by GALNT3 for functionality) or in the FGF23 co-receptor klotho gene. 195 Hyperostosis hyperphosphataemic syndrome is a variant of FTC due to mutation in the GALNT3 gene and is characterized by hyperostosis of long bones. 255
Autosomal dominant hypoparathyroidism is a disorder due to an activating mutation in the calcium sensing receptor (CaSR). As a result, the threshold for suppressing PTH release is decreased and what would be otherwise normal serum calcium is sensed as high and PTH is suppressed. In the renal tubules, activation of CaSR results in an increase in calcium excretion, which would be inappropriate for the prevailing hypo/normocalcaemia. Because of the functional hypoparathyroidism, phosphate excretion is reduced leading to hyperphosphataemia. These patients are often asymptomatic and detected incidentally. 256 Familial isolated hypoparathyroidism is proposed to be caused by a mutation in the preproPTH gene198 or in the glial missing B (GCMB),196 a transcription factor involved in the regulation of PTH both leading to PTH deficiency and raised phosphate. Mutation in the PTH gene has also been described as a possible cause of autosomal recessive hypoparathyroidism. 257
HDR syndrome (hypoparathyroidism, sensorineural deafness and renal abnormalities) is an autosomal disorder caused by mutation in the GATA-3 gene, which encodes a transcription factor. 258
Pseudohypoparathyroidism is a condition characterized by hypocalcaemia and hyperphosphataemia due to resistance to PTH. There are several types: type 1a is due to maternal inheritance of GNAS gene leading to deficiency of the alpha subunit of the GS protein which is involved in coupling of PTH to the cAMP system. These patients may have resistance to other hormones such as TSH, FSH and LH. Type 1b is characterized by resistance to PTH but morphological features seen in type 1a are not present; type 1c is similar to type 1a but deficiency of GS cannot be demonstrated. 197
Consequences of hyperphosphataemia
Acute effects
Acute hyperphosphataemia may cause hypocalcaemia, tetany and hypotension. 259 Hypocalcaemia is due to the precipitation of calcium phosphate. High phosphate will also cause a decrease in the production of 1, 25DHCC by a direct effect as well by the increased FGF23. 260 Soft tissue calcification may occur in many sites including the myocardium, lungs and occasionally the liver. 261 Acute nephropathy and renal failure may result from hyperphosphataemia. 262
Chronic effects
Chronic hyperphosphataemia can affect many tissues particularly the vascular system, kidney, bones, skin, joints and the heart. Serum phosphate has long been recognized as an important risk factor for all-cause and cardiovascular mortality in CKD patients. 263 In recent years, this observation has been extended to the non-CKD population. 264 Serum phosphate has been found to be associated with the severity of coronary atherosclerosis, 264 with increased carotid intima thickness 265 and with left ventricular mass. 266 Possible mechanisms related to the cardiovascular effects of phosphate include vascular calcification, endothelial dysfunction, myocardial hypertrophy and cardiac malfunction. 267 Receptors for phosphate-related hormones have been demonstrated throughout the cardiovascular system 268 and elevated phosphate has been shown to induce vascular calcification. 269 Post-prandial elevation of serum phosphate was shown to impair endothelial function 270 and a consistent positive association has been found between FGF-23 and left ventricular hypertrophy. 271 There are currently no large-scale studies to address the possible causal relationship between phosphate and cardiovascular disease in CKD and non-CKD populations.
Chronic hyperphosphataemia directly or indirectly cause abnormalities in bone. High phosphate causes secondary hyperparathyroidism both by directly acting on the parathyroid gland and indirectly via lowering serum ionized calcium. In addition, high phosphate decreases the production of 1, 25 DHCC directly and indirectly via FGF23. The net result is abnormalities in bone which may result in fractures.
In chronic hyperphosphataemia, calcium phosphate deposition occurs in many tissues including skin, joints, eyes, tendons and blood vessels. Calciphylaxis (calcific uraemic arteriolopathy) is a recognized fatal complication of renal failure and is characterized by calcification in subcutaneous arteries, infarcts in skin and the neighbouring subcutaneous area. It has been reported in about 5% of long-term dialysis patients. 272 The exact pathogenesis is not clear. 273
Investigation of hyperphosphataemia
In investigating hyperphosphataemia, a history, examination and simple tests such as renal function will eliminate many common causes such as renal failure and tumour lysis syndrome. Further investigations are outlined in Figure 4. Measurement of serum PTH is vital because if it is not elevated and the serum calcium is low, hypoparathyroidsm is likely. If PTH is elevated, pseudohypoparathyrodisim should be considered.
274
Investigation of hyperphosphataemia.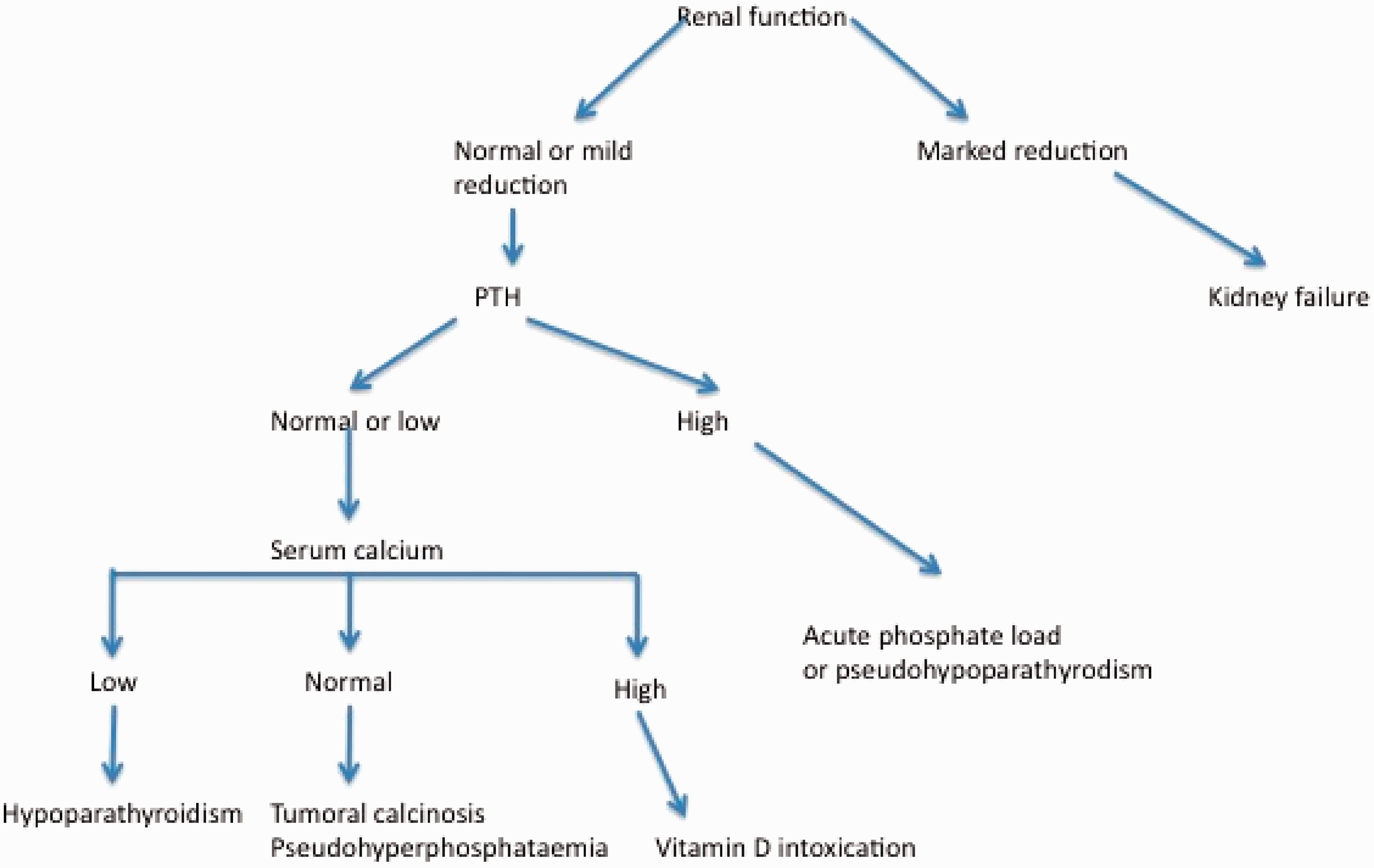
Management of hyperphosphataemia
Principles of management of hyperphosphataemia include determining and treating the underlying cause, reducing the intake of phosphate and increasing the excretion of phosphate. Mild hyperphosphataemia usually does not require treatment; however, in view of the recent suggestions that high phosphate may increase cardiovascular mortality, attempts to reduce phosphate may be advisable.
In situations where hyperphosphataemia is expected, adequate fluid intake and good urine output are essential to minimize the risk. If the cause of the hyperphosphataemia is excessive intake, stopping the supplements is usually adequate. In acute and severe hyperphosphataemia, treatment with intravenous saline and diuretics is helpful to increase the excretion of phosphate if the renal function is adequate. Treatment with insulin and glucose will transiently lower the serum phosphate. Haemodialysis may be required if the renal function is poor.
Patients with CKD should avoid foods such as dairy products, which are high in phosphate. In addition, treatment with phosphate binders is necessary to reduce intestinal absorption. Three classes of phosphate binders are available. The first of these contains aluminium and is no longer recommended due to risk of aluminium toxicity. The second class are calcium-containing binders such as calcium acetate and calcium carbonate. These have the potential side-effects of causing hypercalcaemia and may increase the risk of vascular calcification. The third group of binders are sevelamer and lanthanum carbonate. 275 These are effective in reducing serum phosphate, but a recent study has shown that calcium acetate, lanthanum carbonate and sevelamer carbonate all increase calcification of the coronary arteries and abdominal aorta. 276
Footnotes
Acknowledgements
This article was prepared at the invitation of the Clinical Sciences Reviews Committee of the Association for Clinical Biochemistry and Laboratory Medicine.
Declaration of conflicting interests
None.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethical approval
Not required.
Guarantor
RS.
Contributorship
All authors researched and wrote the review article.