
Abstract
Maturity-onset diabetes of the young (MODY) is a monogenic disorder that results in a familial, young-onset non-insulin dependent form of diabetes, typically presenting in lean young adults before 25 years. Approximately 1% of diabetes has a monogenic cause but this is frequently misdiagnosed as Type 1 or Type 2 diabetes. A correct genetic diagnosis is important as it often leads to improved treatment for those affected with diabetes and enables predictive genetic testing for their asymptomatic relatives. An early diagnosis together with appropriate treatment is essential for reducing the risk of diabetic complications in later life. Mutations in the GCK and HNF1A/4 A genes account for up to 80% of all MODY cases. Mutations in the GCK gene cause a mild, asymptomatic and non-progressive fasting hyperglycaemia from birth usually requiring no treatment. In contrast, mutations in the genes encoding the transcription factors HNF1A and HNF4A cause a progressive insulin secretory defect and hyperglycaemia that can lead to vascular complications. The diabetes in these patients is usually well controlled with sulphonylurea tablets although insulin treatment may be required in later life. In this review, we outline the key clinical and laboratory characteristics of the common and rarer causes of MODY with the aim of raising awareness of this condition amongst health-care scientists.
Introduction
The diagnosis of diabetes for many years has been based on the demonstration of hyperglycaemia. Nearly all classifications have relied on blood (or plasma/serum) glucose concentrations exceeding an established glucose level in a timed sample collection. These include fasting, random relative to prandial status or samples collected after a standardized stress test (such as the 75 g oral glucose tolerance test),1,2 and most recently diabetes diagnosis has been based on HbA1c measurement. 3
Subsequent to the diagnosis of diabetes, patients are categorized into two main subgroups of diabetes, Type 1 or Type 2 diabetes, usually based on clinical criteria such as body mass index (BMI), presence of ketoacidosis and the age of onset. However, it has been known since the first clinical case report by Tattersall4,5 that a discrete group of familial, non-insulin dependent diabetes exists that presents in children and young adults. This clinically heterogeneous group does not fit the classic clinical diagnosis of Type 1 diabetes or Type 2 diabetes and is now known to result from highly penetrant, autosomal dominant mutations in a single gene (monogenic diabetes) leading primarily to β-bell dysfunction. This type of diabetes was coined Maturity Onset Diabetes of the Young (MODY) to reflect the mixed clinical presentation of the juvenile-onset and maturity onset diabetes, terms that were in use at the time.
Advancements in the field of molecular genetics since the first clinical description of MODY have led to the identification of causal mutations in specific genes involved in β-cell function. The subsequent re-classification of MODY based on genetic aetiology has resulted in the recognition of distinct clinical phenotypes with varying features such as age of onset, response to treatment, extra-pancreatic features, severity of hyperglycaemia and subsequently complications and prognosis. 6 The latest guidelines on classification of diabetes published by the American Diabetes Association and the World Health Organisation recognizes these genetic subtypes.1,2
Heterozygous mutations in the GCK and HNF1A/4 A genes account for up to 80% of all MODY cases. 7 GCK encodes the intra-cellular enzyme glucokinase (GCK), which acts as a glucose sensor in the pancreatic β-cells. Mutations in GCK result in a mild, often asymptomatic and non-progressive fasting hyperglycaemia requiring no treatment, except perhaps during pregnancy.8,9 In contrast, mutations in the genes which encode the transcription factors hepatocyte nuclear factor-1 alpha and −4 alpha (HNF1A and HNF4A) cause a progressive insulin secretory defect and hyperglycaemia that can lead to the development of vascular diabetic complications. 10 The diabetes in these patients is often initially well controlled using sulphonylurea tablets although insulin treatment may be required in later life. 11 Diabetes resulting from mutations in another transcription factor gene, HNF1B, is much rarer but is associated with extra pancreatic manifestations, the most common being renal cysts. 12 At least 10 discrete genetic aetiologies of MODY have been described to date.
The prevalence of MODY is rare and is estimated to account for between 0.6% and 2% of all diabetes.13–15 No population-based study has been performed and MODY is often misclassified as Type 1 or Type 2 diabetes, so prevalence is likely to be underestimated. Even if the minimum reported prevalence is assumed, then in the UK more than 80% of MODY has not been diagnosed by molecular genetic testing. This means that thousands of patients are not receiving the most appropriate treatment and their unaffected relatives are unaware of their increased risk of diabetes. In addition, there has been 10-fold regional variation in the reported prevalence of confirmed MODY. This variation is the direct result of differences in referral rates, most probably reflecting variable awareness of MODY across the country or limited access to molecular genetic testing. 13
Differentiating maturity onset diabetes of the young from Type 1 and Type 2 diabetes.
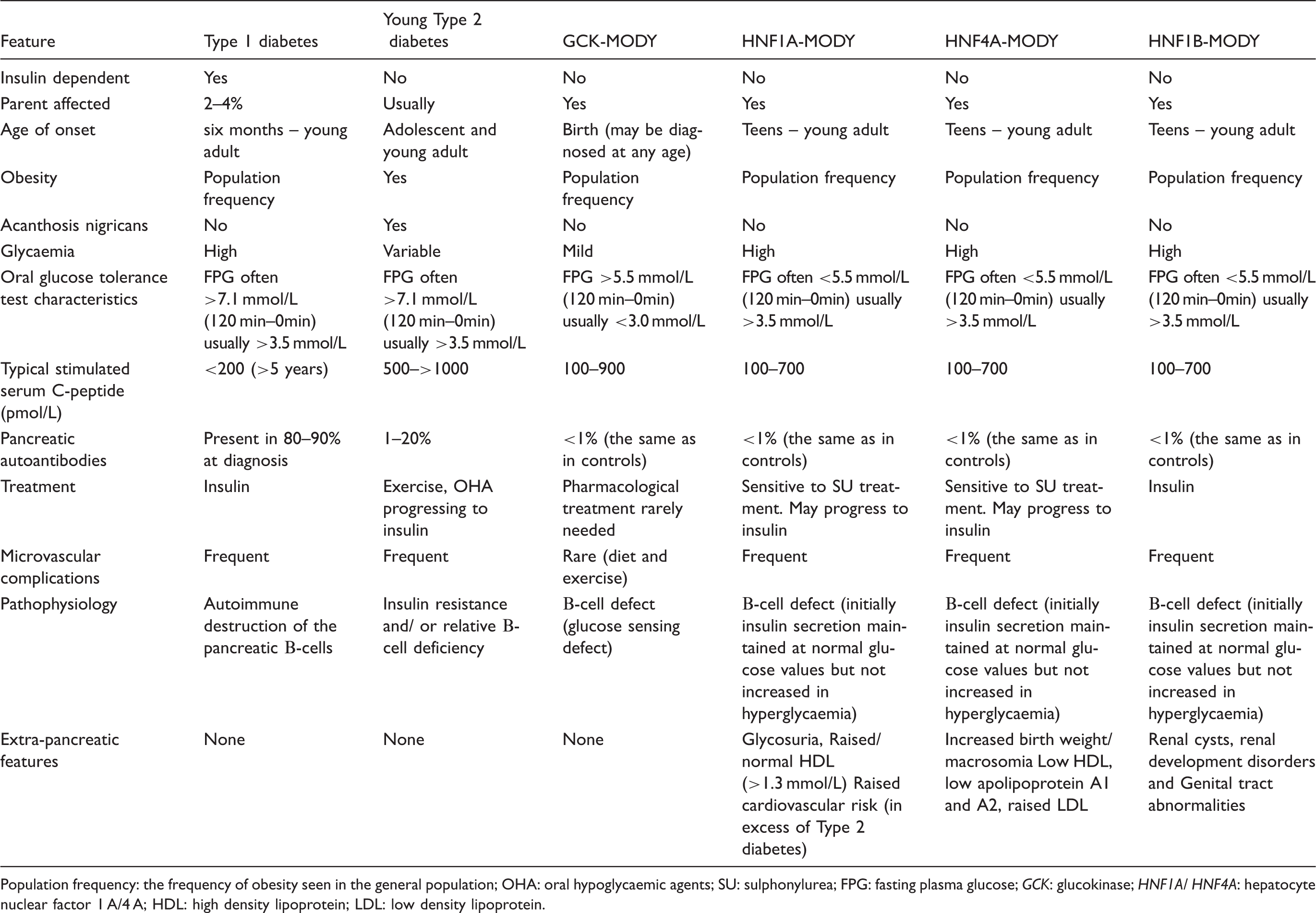
Population frequency: the frequency of obesity seen in the general population; OHA: oral hypoglycaemic agents; SU: sulphonylurea; FPG: fasting plasma glucose; GCK: glucokinase; HNF1A/ HNF4A: hepatocyte nuclear factor 1 A/4 A; HDL: high density lipoprotein; LDL: low density lipoprotein.
In this review, we will outline the key characteristics of Maturity Onset Diabetes of the Young and raise awareness of basic laboratory tests and clinical characteristics that can serve as useful tools to help identify MODY patients. In doing so we highlight the importance of a correct genetic diagnosis in these patients and emphasize that health-care scientists are in a unique position to help identify misdiagnosed and undiagnosed MODY mutation carriers.
GCK-MODY
Inactivating heterozygous mutations in the GCK gene results in GCK-MODY which is characterized by a mild fasting hyperglycaemia from birth, typically in the range of 5.5–8.5 mmol/L, with only marginal deterioration in glycaemic control seen in individuals with increasing age.6,17
Pathophysiology of GCK-MODY
GCK is the enzyme involved in the first step of glucose metabolism and catalyses the transfer of a phosphate from ATP to glucose, generating glucose-6-phosphate in both hepatocytes and pancreatic β-cells (Figure 1). This is the rate limiting step in glucose metabolism and the activity of GCK is directly proportional to the ambient glucose concentration. As such, in the β-cells, GCK serves as a ‘glucose sensor’ facilitating insulin release that is both appropriate and proportional to the blood glucose concentration.
18
Schematic representation of glucose-induced insulin secretion. Glucose enters the β-cell via the GLUT2 transporter. Once inside the cell glucokinase phosphorylates glucose to glucose-6-phosphate (1). Glucose-6-phosphate enters the glycolytic pathway and is metabolized to pyruvate. Pyruvate enters the mitochondrial citric acid cycle with a net production of ATP, reciprocal fall MgADP (2). An increase ATP/MgATP ratio acts to close the KATP channel (3) provoking membrane depolarization, opening of the voltage-gated Ca2+ channels (4), Opening of voltage gated calcium channels increases calcium influx into the cell (5), which is the trigger for insulin exocytosis (6).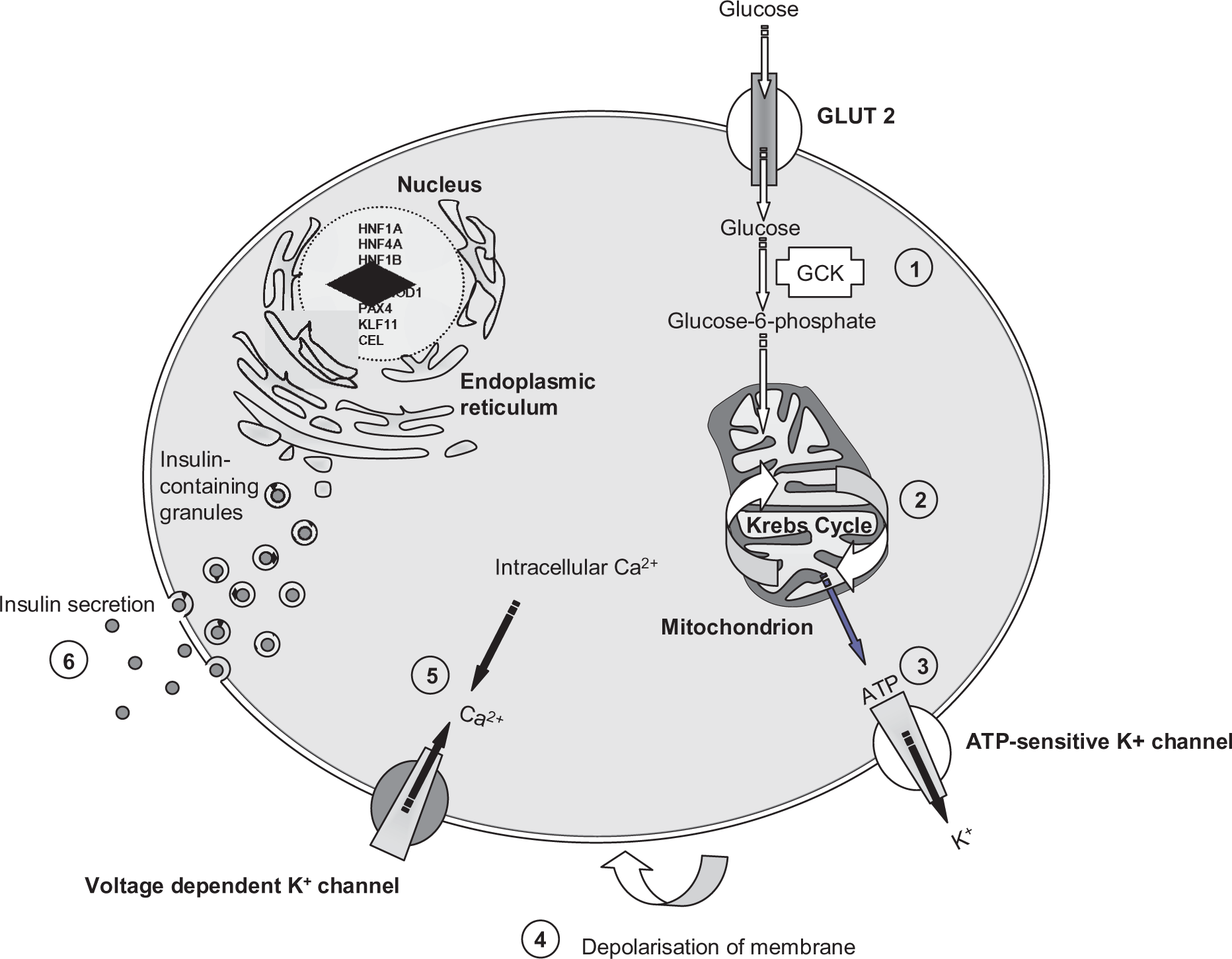
Heterozygous loss-of-function mutations in GCK result in a decreased rate of phosphorylation and the insulin dose–response curve is shifted to the right. 19 As a result the glycaemic threshold for insulin release is regulated at a slightly higher set-point but still remains under tight homeostatic control. This mechanism for hyperglycaemia in GCK-MODY is supported by the observation that GCK mutation carriers have a reduced insulin secretion at a given glucose concentration compared with controls. 19 GCK is also expressed in the liver and patients with GCK-MODY have reduced hepatic glycogen synthesis and stores. 20
Over 600 loss-of-function mutations have been identified throughout the 10 exons and promoter of the pancreatic β-cell GCK gene and all cause a remarkably similar phenotype due to compensatory activity of the unaffected GCK allele. 21
Interestingly, in contrast to the heterozygous loss-of-function mutations in GCK-MODY, homozygous loss-of-function GCK mutations result in complete deficiency of this enzyme and are a rare cause of permanent insulin-requiring diabetes presenting in the neonatal period. 22 Heterozygous gain-of-function mutations cause the opposite phenotype of hyperinsulinaemic hypoglycaemia. 23
Clinical features of GCK-MODY (
Table 1
)
The mild hyperglycaemia in GCK-MODY is asymptomatic and postprandial glucose excursions are modest, with a 75 g oral glucose tolerance test increment less than 3 mmol/L in 70% of patients 17 and 4.6 mmol/L in 95% (Ellard and Hattersley, unpublished data). HbA1c values are consequently near normal and microvascular and macrovascular complications are rare. 24 Because the hyperglycaemia of GCK-MODY is mild, many GCK patients are detected incidentally at routine screening for an unrelated illness or during pregnancy. For the same reason affected family members may not have been diagnosed and therefore the autosomal dominant pattern of inheritance may not be evident. Testing apparently unaffected parents by measuring fasting glucose often reveals that one parent has mild hyperglycaemia.
A large proportion of all incidental persistent mild hyperglycaemia seen in non-obese young adults has been shown to be attributable to GCK-MODY (up to 43%). 25 The diagnosis of GCK-MODY is important in this group as they may be assumed to be developing Type 1 diabetes or have Type 2 diabetes and can receive treatment that is unnecessary and ineffective. 25
The identification of a GCK mutation in women during pregnancy has an additional significance. If a baby does not inherit a GCK mutation from its affected mother it will be at risk of macrosomia as a result of increased insulin secretion and insulin-mediated foetal growth secondary to maternal hyperglycaemia. 26 If a baby inherits an inactivating GCK heterozygous mutation from the father the birth weight is reduced by approximately 500 g as a result of reduced foetal insulin. 27 In addition, management of GCK-MODY both during and outside pregnancy is different compared to other subjects with gestational diabetes. 28 Outside pregnancy, hypoglycaemic medication is not recommended for patients with GCK-MODY as hyperglycaemia is mild, complications are rare and medication has been shown to have minimal effect. 29
How to recognize GCK-MODY from laboratory test results and key clinical features
The following features are suggestive of GCK-MODY:
Persistent fasting hyperglycaemia in the range 5.5–8.5 mmol/L An oral glucose tolerance test glucose increment (120 min glucose minus 0 min glucose) of <3.0 mmol/L Negative pancreatic autoantibodies (the prevalence of pancreatic autoantibodies in MODY is the same as in controls)
30
A near normal HbA1c (values > 55 mmol/mol [7.5%] would be suggestive of an alternative diagnosis) Persistent fasting C-peptide production (stimulated serum C-peptide >200 pmol/L) (Table 1) A parent will usually have mild fasting hyperglycaemia (5.5–8.5 mmol/L)
Transcription factor MODY
HNF1A-MODY
In contrast to GCK-MODY, heterozygous mutations in the HNF1A gene encoding the transcription factor hepatocyte nuclear factor-1 alpha (HNF1A) cause autosomal dominant diabetes that is associated with long-term diabetic complications.
Pathophysiology of HNF1A-MODY
HNF1A, along with HNF4A and HNF1B constitute part of a network of transcription factors expressed in several tissues in the body that interact to orchestrate gene expression during embryonic development. In the mature β-cell the hepatic nuclear factor transcription factors also regulate the expression of insulin as well as altering β-cell development, proliferation and cell death.
Mutations in these transcription factors have been shown to alter gene expression for proteins involved in glucose transport (including the glucose transporter GLUT 2), glucose metabolism and key enzymes in mitochondrial glucose metabolism.31–33 Reduced β-cell proliferation and increased apoptosis have been proposed to explain the progressive decline in β-cell function that is characteristic in these patients.33–36
The penetrance of HNF1A mutations is high, with 63% of carriers developing diabetes by 25 years of age, 79% by 35 years and 96% by 55 years. 37 The location of the mutation within the HNF1A gene is a factor in determining the age of diabetes onset. 38
Clinical features of HNF1A-MODY
The diabetes in HNF1A-MODY typically presents in adolescence or early adulthood before the age of 25 years.39,40 These patients are born with normal glycaemia, tend to be slim and have normal insulin sensitivity. Diabetes develops in HNF1A-MODY as a result of progressive failure of insulin secretion.
Microvascular and macrovascular complications are observed in HNF1A-MODY and are related to poor glycaemic control. Patients show deteriorating glycaemic control with age and microvascular complications occur with the same frequency as in patients with Type 1 and Type 2 diabetes. 41 In a 75 g oral glucose tolerance test patients tend to show a large glucose increment (>5 mmol/L). Interestingly, HNF1A-MODY patients often have a normal fasting glucose concentration. This is believed to be because in early stages of diabetes they maintain sufficient insulin secretory capacity to achieve euglycaemia in the fasting state; this is aided by relative insulin sensitivity and low BMI. 17
HNF1A extra-pancreatic features
Patients with HNF1A-MODY have some interesting extra-pancreatic features reflecting that the HNF1A gene is expressed in tissues outside the pancreas (Table 1). These patients have glycosuria because of a low renal threshold for glucose, thought to be due to reduced expression of the sodium-glucose cotransporter 2 (SGLT-2) and reduced glucose reabsorption in the proximal tubule. 42 In non-diabetic mutation carriers, glycosuria can be detected in approximately 38% of patients after an oral glucose tolerance test even when blood glucose concentrations remain in the reference range. 43
In addition, serum lipoprotein analysis in these patients demonstrates a higher than normal high density lipoprotein (HDL) cholesterol concentration which is in contrast to the low concentration that is normally observed in Type 2 diabetes and normal concentrations seen in Type 1 diabetes. 44 This observation may be predicted to decrease cardiovascular risk, but despite the high HDL cholesterol, incidence of coronary heart disease is greater in HNF1A-MODY than in patients with Type 1 diabetes but less than those with Type 2 diabetes. 41
HNF1A treatment response
Patients with HNF1A mutations show marked sensitivity to the oral hypoglycaemic agent sulphonylurea. HNF1A-MODY patients demonstrate a five-fold greater response to sulphonylurea than to standard metformin treatment, whereas in Type 2 diabetes the efficacy of the two drugs has been shown to be equivalent. 45 Better glycaemic control is often achieved with sulphonylurea than on insulin therapy and the fasting glucose lowering effect is four times greater than that seen in BMI and glycaemia matched Type 2 diabetes patients.46,47 As a result of this finding, many patients with HNF1A mutations have successfully transferred from insulin therapy to sulphonylurea tablets with no deterioration in glycaemic control. 48 Even patients receiving insulin treatment for several decades have been able to transfer to sulphonylurea tablets without ketoacidosis.11,48 Switching from injectable insulin therapy to tablet sulphonylurea treatment is successful in the majority of patients and has a major impact on the quality of life for these patients.11,49
Low-dose sulphonylurea should be the first-line treatment choice in HNF1A-MODY, although as diabetes progresses the addition of insulin therapy may be required in some patients. 11
How to recognize HNF1A-MODY from laboratory test results and key clinical features
The following features are suggestive of HNF1A-MODY:
An oral glucose tolerance test glucose increment (120 min glucose minus 0 min glucose) of >5.0 mmol/L Negative pancreatic autoantibodies (the prevalence of pancreatic autoantibodies in MODY is the same as in controls) Persistent fasting C-peptide production (stimulated serum C-peptide >200 pmol/L) (Table 1) Normal/raised HDL cholesterol (>1.3 mmol/L) Glycosuria at blood glucose <10.0 mmol/L
HNF4A-MODY
Pathophysiology and clinical features of HNF4A-MODY
Mutations in the HNF4A gene causing MODY are less common than HNF1A-MODY and account for approximately 3–5% of MODY cases. 50 The diabetes in HNF4A-MODY presents in a similar way to HNF1A-MODY and patients have a progressive β-cell dysfunction. This is thought to be because in adults both HNF1A and HNF4A act within the same transcription pathway. 10
The penetrance of HNF4A gene mutations is variable and the majority of mutation carriers develop diabetes by the age of 25 years, but some affected individuals still have not developed diabetes in their fourth decade. Sensitivity to sulphonylureas is also characteristic of HNF4A mutations and low-dose sulphonylurea therapy should be the considered as first-line treatment. 45
The identification of an HNF4A mutation has important implications for the management of pregnancy. Mutations in HNF4A are associated with an 800 g increase in birth weight compared to unaffected siblings. 51 Therefore, the baby of an affected mother or father has a 50% risk of developing macrosomia and should be monitored closely.
In addition, heterozygous HNF4A mutations cause diazoxide-responsive hyperinsulinaemic hypoglycaemia presenting in the first week of life in ∼10% of mutation carriers.51,52
HNF4A-MODY extra-pancreatic features
Unlike HNF1A-MODY, patients with HNF4A mutations have reduced concentrations of total HDL-cholesterol, apolipoprotein A1, apolipoprotein A2 and triglycerides and frequently have raised LDL-cholesterol concentrations.45,53
How to recognize HNF4A-MODY from laboratory test results and key clinical features
The following features are suggestive of HNF4A-MODY:
An oral glucose tolerance test glucose increment (120 min glucose minus 0 min glucose) of >5.0 mmol/L Negative pancreatic autoantibodies (The prevalence of pancreatic autoantibodies in MODY is the same as in controls) Persistent fasting C-peptide production [stimulated serum C-peptide >200 pmol/L] (Table 1) Low HDL cholesterol, low triglyceride and raised LDL cholesterol Macrosomia and/or congenital hypoglycaemic hyperinsulinaemia
Rare forms of MODY
HNF1B-MODY (renal cysts and diabetes syndrome)
Mutations in the gene encoding the transcription HNF1B factor expressed in the pancreas, kidneys, liver, genital tract and gut are a rare but phenotypically distinct cause of MODY. 54
Pathophysiology and clinical features of HNF1B-MODY
Mutations in the HNF1B gene account for approximately 1% of all MODY cases. 55 Unlike HNF1A- and HNF4A-MODY where the predominant feature is β-cell dysfunction, the diabetes that develops in approximately half of HNF1B mutation carriers is the result of both β-cell dysfunction and insulin resistance. The sensitivity to sulphonylureas associated with HNF1A and HNF4A mutations is absent, and early insulin therapy is required. 56 The penetrance of HNF1B-MODY is highly variable, with diabetes diagnosed from 0–61 years.57,58 Approximately one-third of HNF1B mutations are deletions of the entire gene and genetic testing for HNF1B should include a method of dosage analysis to identify such deletions. 59 De novo mutations are frequent (up to 50% of cases) and hence family history may be absent.57,58,60–62
Low birth weight (reduction of 800 g) is common in babies carrying an HNF1B mutation as a result of reduced insulin secretion in utero. 58 HNF1B mutations causing transient neonatal diabetes (TNDM) have been reported. 58
HNF1B-MODY extra-pancreatic features
The HNF1B transcription factor is expressed in early embryonic development in the pancreas, kidney, liver and genital tract and in HNF1B mutation carriers developmental abnormalities can be detected in all these organs. The predominant extra-pancreatic feature of HNF1B-MODY is the development of renal disease resulting in a clinical syndrome of Renal Cysts and Diabetes (RCAD). The most common abnormality is renal cysts but renal dysplasia and renal-tract malformations such as horse shoe kidney and/or familial hypoplastic glomerulocystic kidney disease have been reported.58,63,64
Approximately 50% of subjects will develop end-stage renal failure before the age of 45 years, requiring renal replacement and fewer than 6% known HNF1B-MODY patients have normal renal function. 57 Genital tract abnormalities are found in some cases, but as with all the phenotypic features of HNF1B-MODY the penetrance is incomplete. Malformations include vaginal aplasia, rudimentary uterus, bicornuate uterus and double vagina.65–67 Other features of HNF1B-MODY include hyperuricaemia (and associated gout), abnormal liver function tests and hypomagnesaemia.68–70
How to recognize HNF1B-MODY from laboratory test results and key clinical features
An oral glucose tolerance test glucose increment (120 min glucose minus 0 min glucose) of >5.0 mmol/L Negative pancreatic autoantibodies (The prevalence of pancreatic autoantibodies in MODY is the same as in controls) Persistent fasting C-peptide production [stimulated serum C-peptide >200 pmol/L] (Table 1) Renal impairment with raised creatinine Hypomagnesaemia Hyperuricaemia Raised liver enzymes
Rare forms of MODY
Mutations in HNF1A, HNF4A, HNF1B and GCK genes account for over 80% of all known MODY cases. However, mutations in other genes are known to be rare causes of autosomal dominant diabetes including IPF1, NEUROD1, CEL, INS, KCNJ11 and ABCC8.
IPF1-MODY
Insulin promoter factor 1 is a pancreatic transcription factor and like the HNF-family of transcription factors regulates β-cell development and insulin gene expression. The first case of MODY caused by a heterozygous mutation in the IPF1 (PDX1) gene was discovered in 1997.71,72 Since then no further MODY cases have been reported, suggesting it is a very rare cause of MODY.71,73,74 However, recessive mutations causing neonatal diabetes have been identified in two further families71,75,76 and a history consistent with MODY was reported in one.71,75,76
NEUROD1-MODY
Heterozygous mutations in NEUROD1, which encodes the basic helix-loop-helix transcription factor Neurogenic Differentiation 1, are another very rare cause of MODY. 77 The NEUROD1 gene is involved in neuronal differentiation, development of endocrine cell lineages and alters transcription of GLUT2, insulin and GCK.78,79 To date only five families have been reported with heterozygous mutations in the NEUROD1 gene causing MODY.80–82
Recently, two different homozygous mutations in NEUROD1 were shown to be responsible for a novel syndrome of permanent neonatal diabetes and neurological abnormalities. 83
CEL-MODY
The CEL gene encodes the enzyme carboxyl ester lipase (bile salt-stimulated lipase) that is secreted from the pancreas into the digestive tract. Its physiological role is in cholesterol and lipid-soluble vitamin ester hydrolysis and absorption from the intestine.
In 2006, two Norwegian families with autosomal dominant young onset diabetes were found to harbour mutations in the CEL gene. 84 The affected subjects were also found to have faecal elastase deficiency and other evidence of pancreatic exocrine dysfunction.
Both families demonstrated a different single-base deletion causing a frameshift in the variable number of tandem repeats of the gene.84,85 One of these families included 14 diabetic members and the logarithm (base 10) of odds (LOD) score for linkage with diabetes was 4.5 (where ≥3 is significant). In one study pancreatic enzyme substitution alleviated fat malabsorption but glycaemic control was not affected. 86 To date only three families have been identified with CEL mutations causing MODY.84,85
INS-MODY
Dominant mis-folding mutations in the INS gene are the second most common cause of isolated permanent neonatal diabetes (PNDM) with variable age at onset of diabetes.87,88 Six families from Norway, Denmark, France, Czech Republic and the UK have thus far been reported to have heterozygous INS missense mutations that co-segregate with MODY.87,89–91
Potassium channel (KATP) MODY
The ABCC8 and KCNJ11 genes encode the SUR1 and Kir6.2 subunits of the ATP-sensitive potassium (KATP) channel in the pancreatic β-cell which has a direct role in regulating insulin release (Figure 1).
Dominant and recessive activating mutations in the KATP channel genes cause neonatal diabetes presenting in the first six months of life which can be permanent (PNDM) or transient (TNDM).92–94 Both forms are responsive to high-dose sulphonylurea therapy.95,96 In contrast, recessive loss-of-function mutations in ABCC8 and KCNJ11 are the most common cause of congenital hypoglycaemic hyperinsulinism (CHI). 97
The milder activating mutations that cause transient neonatal diabetes may be inherited from a parent who was diagnosed with permanent diabetes outside the neonatal period.93,98,99 ABCC8 and KCNJ11 mutations may therefore be classified as a rare cause of MODY.94,100
Other potential forms of MODY
Mutations in the genes encoding a further three transcription factors, KLF11, PAX4 and BLK, have been reported to cause MODY in two (KLF11) 101 or three families (PAX4102,103and BLK 104 ), but the combined LOD scores for each gene do not meet statistical significance. Identification of mutations in additional pedigrees is required to confirm these as ‘MODY genes’.
Genetic testing
Genetic testing for MODY is offered by specialist centres around the world (see www.diabetesgenes.org for details of the UK centre). Sanger DNA sequencing is the gold standard mutation screening method although other methods are sometimes used (e.g. denaturing high-performance liquid chromatography [dHPLC]).
Partial and/or whole gene deletions are estimated to represent up to 3% of all MODY gene mutations but these cannot be detected by sequencing. 105 Gene dosage analysis using Multiplex Ligation-dependent Probe Amplification (MLPA) can be used to detect the presence of these gene deletions, and the assay is typically employed where a diagnosis of MODY is strongly suspected but no mutation is found by DNA sequencing. The European Molecular Genetic Quality Network (www.emqn.org) runs an annual quality assurance scheme for monogenic diabetes and has published Best Practice Guidelines for molecular genetic testing in monogenic diabetes. 7
Biomarkers to identify MODY
There has been a drive to identify sensitive, specific, cheap and widely available biomarkers that are good at discriminating MODY from other forms of diabetes. Biomarkers proposed for the discrimination of HNF1A or HNF4A MODY from Type 2 diabetes include 1,5-anhydroglucitol, complement factors C5 and C8, apolipoprotein M and transthyretin, but none has sufficient sensitivity and specificity, 106–110 or the required reproducibility to be clinically useful.109,111
One marker that has shown more promise is serum high sensitivity C-reactive protein which is significantly lower in HNF1A-MODY than in Type 1 diabetes, Type 2 diabetes, GCK-MODY and non-diabetic controls. 112 The CRP gene has HNF1A binding sites in its promoter and common variants in and around HNF1A are associated with circulating high sensitivity C-reactive protein (hsCRP) concentrations.113–115 Importantly, the differences observed are large enough that the sensitivity and specificity of hsCRP at discriminating HNF1A-MODY from Type 2 diabetes are >70%, making it potentially a useful clinical test.
Recently, efforts have been made to formally assess the discriminative ability of traditional diabetes laboratory tests such as C-peptide production and pancreatic autoantibodies. Persistent postprandial C-peptide production, assessed in a spot urine sample, has a sensitivity and specificity of >95% at discriminating HNF1A- and HNF4A-MODY from long duration Type 1 diabetes. 116 The absence of the pancreatic autoantibodies GAD and/or IA2 has been shown to discriminate Type 1 diabetes from HNF1A, HNF4A and GCK MODY with a sensitivity of 99% and specificity of 84%. 30 These tests are widely available and may provide cost-effective screening tools to support more precise targeting of MODY diagnostic testing.
Summary
A correct genetic diagnosis of monogenic diabetes is important for optimizing patient treatment choices, with some patients being able to come off insulin and switch to sulphonylurea tablets (HNF1A/4A-MODY) and others requiring no treatment at all (GCK-MODY). A diagnosis of MODY is also important for predicting course of disease, extra-pancreatic phenotype and also enables predictive testing for first degree relatives who are at 50% risk of inheriting the mutation.
Despite the fact that 80% of MODY patients in the UK remain unidentified, genetic testing is too costly and labour intensive to implement a universal screening programme for all patients with diabetes. A rapid advance in the field of molecular genetics has seen the introduction of new high capacity sequencing technologies and with it the expectation of decreasing genetic test costs. This may lead to a future where it is possible to perform gene analysis in all patients diagnosed with young onset diabetes.
The identification and characterization of inexpensive and universally available biomarkers may help health-care professionals identify appropriate patients for genetic screening. This would increase diagnosis rates for MODY by identifying those patients more likely to have a monogenic form of diabetes rather than Type 1 or Type 2 diabetes.
Health-care scientists are uniquely placed to educate clinicians and facilitate identification of these patients. The ultimate challenge is to translate research discoveries into improved clinical practice and quality of life. This requires an early, accurate genetic diagnosis and implementation of appropriate treatment leading to considerable lifetime savings in drug therapy, reduced blood glucose monitoring, reduced clinical follow-up and better glycaemic control/early diagnosis in relatives, leading to a lower frequency of diabetic complications.
Footnotes
Declaration of conflicting interests
None.
Funding
TM and SE are supported by the European Community FP7 program CEED3 (HEALTH-F2-2008-223211) and the Health Innovation Challenge Fund (HICF-1009-0041). SE is supported by the NIHR Peninsula Clinical Research Facility, University of Exeter.
Ethical approval
N/A.
Guarantor
SE.
Contributorship
Tim McDonald and Sian Ellard co-wrote and edited the manuscript.