
Abstract
The identification of stroke cases caused by monogenic disorders is important both for therapeutic decisions and genetic counselling, although they represent less than 1% of all stroke patients. The purpose of this review is to summarize genetic, pathological, and clinical features of single-gene disorders related to ischemic stroke. The following monogenic disorders are considered: cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy, cerebral autosomal-recessive arteriosclerosis with subcortical infarcts and leukoencephalopathy, hereditary endotheliopathy with retinopathy, nephropathy, and stroke, Fabry disease, pseudoxanthoma elasticum, Neurofibromatosis type 1, familial MoyaMoya disease, Ehlers-Danlos syndrome type IV, Marfan syndrome. For each monogenic disorder, mode of inheritance, pathophysiological aspects, clinical phenotype, and diagnostic tools are carefully described. Furthermore, the classification of monogenetic disorders is presented according to stroke mechanisms, which include small vessel diseases, large artery diseases, and arterial dissections. This review could be useful to identify specific diagnostic pathways for patients with a suspicion of monogenic disease.
Introduction
Stroke is a multifactorial and polygenic disease (Hassan and Markus, 2000; Rubattu et al, 2000), which has a monogenic basis in a small percentage of cases. The identification of a genetic abnormality may have relevant implications for therapeutic management and genetic counseling; therefore, it is important to do the diagnosis. Although the young age at stroke onset, the absence of conventional risk factors, and a family stroke history can suggest the presence of a single-gene disorder, it can be difficult for a physician to decide which stroke patients to screen for a monogenic disorder and for which monogenic disorder to screen them. A large number of single-gene disorders such as hypercoagulable states, nonatherosclerotic vasculopathies and cardiac, hematologic, or connective disorders have been described as well-known causes of stroke. In the literature, there is no standard classification for monogenic stroke syndromes, and a single-gene disorder can be associated with different stroke phenotypes (cardioembolic, small vessel, and large vessel). In this article, we review monogenic stroke disorders related, above all, to small and large arterial abnormalities leading to stenosis, occlusion, and dissection of blood vessels. The purpose of this paper is to summarize, for each single-gene disorder associated with ischemic stroke, an updated description of mode of inheritance, pathophysiological aspects, clinical phenotype, and diagnostic pathway. This report could represent a useful tool for the clinician seeing stroke patients to perform the correct diagnosis.
Methods
Search Strategy and Study Selection
References for this review were identified consulting electronic databases (MEDLINE and EMBASE) until July 2006 and references of relevant articles. A wide number of articles were also identified through searches of the extensive files of the authors. The search results were limited to monogenic disorders leading to vessel disease and therefore ischemic stroke. The search terms ‘cerebrovascular disorder’ (OR) ‘ischemic stroke’ combined with the text words ‘genetic’ (OR) ‘monogenic’ (OR) ‘single-gene’ were used. For each disease, the articles related to epidemiology, genetic analysis, pathology, clinical phenotype, and diagnosis were selected, and the full papers were evaluated. We included in our analysis abstracts, letters, articles, case control studies, prospective cohort studies, and reviews on specific diseases, not just papers published in English were reviewed.
Small vessel diseases
Cerebral Autosomal-Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy
Cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is a generalized disease of the small arteries, largely predominating in the brain (Tournier-Lasserve et al, 1993).
Epidemiology: Varying phenotypic expressions of CADASIL lead to underrecognition, and its prevalence, therefore, remains uncertain. However, Razvi et al (2005) estimated the minimum prevalence of CADASIL (1.98/100.000) in the Scottish population.
Genetic and Pathological Aspects: Joutel et al (1996) identified Notch 3, on chromosome 19, as the responsible gene of CADASIL. The types of mutations identified were missense mutations (Dichgans et al, 2000), splice-site mutations (Joutel et al, 2000), and small in frame deletions (Dichgans et al, 2000). Notch 3 gene codes a transmembrane receptor containing a large number of tandemly repeated epidermal growth factor-like domains (Artavanis-Tsakonas et al, 1995). The Notch3 is involved in cell fate specification during embryonic development, but the role in normal adult cells is unknown. How this mutated gene can lead to the CADASIL phenotype is still unclear. The pathological pattern of CADASIL is a nonatherosclerotic, amyloid-negative angiopathy involving small arteries. Histopathological studies discovered deposits of granular eosinophilic material (periodic acid-Schiff)-positive, called ‘granular osmiophilic materials’ (GOM), in the vascular smooth muscle cells of small arteries and arterioles. The vascular alterations may involve the peripheral nerve, skin, muscle, retina, and visceral organs, but the brain is preferentially involved (Ruchoux and Maurage, 1997). Granular osmiophilic material deposits can be detected on electron microscopy in direct contact with vascular smooth muscle cells (Arima et al, 2003; Ruchoux and Maurage, 1997). The origin and structure of GOMs is unknown, but they are pathognomonic of the disease. The skin vessel morphometry shows an early and widespread destruction of vascular smooth muscle cells with loss of extracellular matrix, leading to a loss of cement between the different cells of the vessel wall (Brulin et al, 2002).
Clinical Phenotype: The clinical phenotype is highly variable even within single families and is characterized by recurrent ischemic episodes (transient ischemic attack (TIA) or stroke), attacks of migraine with aura, mood disorders, cognitive deficits, and epileptic seizure (Chabriat et al, 2000; Dichgans et al, 1998).
Stroke or TIA occurs in 84% of cases (Chabriat et al, 1995), and mean age at onset is around 46 years (range: 30 to 66) (Dichgans et al, 1998). Most of these episodes are lacunar syndromes that occur in the absence of normal vascular risk factors; instead cortical infarcts are very rare (Rubio et al, 1997). The recurrent ischemic episodes lead to progressive disability with urinary incontinence or pseudobulbar palsy (Opherk et al, 2004). Cognitive decline, documented in 48% of cases (Dichgans et al, 1998), includes subcortical dementia with frontal-like symptoms (poor attention, perseveration, and apathy) and deficit in memory. Dementia is multiinfarct with a slow progression and a stepwise deterioration (Chabriat et al, 1995). The migraine with aura is present in approximately 35% of cases (Davous, 1998) and is among the earliest manifestations of the disease. The aura symptoms, which may be visual or somatosensory are difficult to distinguish from an ischemic episode (Chabriat et al, 1995). Psychiatric disorders were observed in approximately 30% of patients, and mood disorders are the most frequent diagnoses (Dichgans et al, 1998). Epilepsy is infrequently reported (6 to 10%) and its pathophysiology is unclear (Davous, 1998).
Diagnosis
Neuroimaging: Magnetic resonance imaging (MRI) is useful in making a diagnosis. It shows areas of hyperintensity of white matter easily detected by T2-weighted MRI and lacunar infarcts that involve lobar white matter, external and internal capsule, basal ganglia, and thalamus. These images suggest an involvement of small arteries mainly affecting the perforating vessel of the brain (Chabriat et al, 1995, 1998). Similar lesions are observed on a sporadic basis in patients with cardiovascular risk factors, especially hypertension, in the absence of Notch 3 mutations. A more specific MRI feature of CADASIL is a diffuse hyperintensity confined to the anterior temporal pole; this abnormality was not seen in patients affected by ischemic leukoaraiosis (O'Sullivan et al, 2001). Some MRI studies have shown a multiplicity of microbleeds in individuals with CADASIL (Dichgans et al, 2002; Lesnik Oberstein et al, 2001), and imaging abnormalities have also been found in asymptomatic patients of affected pedigrees (van den Boom et al, 2003).
Skin Biopsy: Skin biopsy with ordinary electron microscopy can reveal the characteristic deposition of GOM around the vascular smooth muscle cells. This pathological finding is very specific, but its sensitivity varies widely according to the studies (Brulin et al, 2002; Markus et al, 2002). Instead, the immunohistochemical detection of the Notch3 ectodomain in skin biopsy has high 90 to 96% sensitivity and is 95 to 100% specific for diagnosis of CADASIL (Joutel et al, 2001).
Genetic Analysis: The presence of mutations in Notch3 gene confirms the diagnosis. As the Notch3 gene has 33 exons, a complete gene mutation analysis is unsuitable for routine diagnosis and screening strategies have been suggested.
A study in Caucasian patients revealed strong clustering mutations around exons 3 and 4 (65%) and suggested sequencing of these exons as the first step (Joutel et al, 1997). However, other studies showed divergence from this pattern, for example, Dotti et al (2005) screened for mutations in 28 unrelated Italian CADASIL families and found the highest rate of mutations in exon 11. The mutation pattern is largely dependent on the population screened, and it is not possible to suggest an effective screening strategy everywhere.
Treatment: There is no specific treatment for this disease. Therefore, medical treatment is largely aimed at the alleviation of symptoms.
Cerebral Autosomal-Recessive Arteriopathy with Subcortical Infarcts and Leukoencephalopathy
Cerebral autosomal-recessive arteriopathy with subcortical infarcts and leukoencephalopathy is a cerebral small vessel disease with an autosomal-recessive pattern of inheritance.
Epidemiology: It has been described in Japan and only few tens of patients have been reported (Fukutake and Hirayama, 1995).
Genetic and Pathological Aspects: Consanguinity is frequently seen in the patients' parents and the disease seems to be transmitted in an autosomal-recessive manner. The responsible gene is unknown and the pathogenetic mechanism of this disorder remains obscure. Atherosclerotic changes with fibrous intimal thickening and narrowing of the lumen have been described in the vessels of cerebral white matter and basal ganglia. In contrast with CADASIL, periodic acid-Schiff-positive GOM deposition, in the media of small arteries and arterioles, was not found in this disease (Arima et al, 2003).
Clinical Phenotype: About half of the patients had ischemic strokes or stepwise deterioration leading prematurely to vascular dementia. The onset of cerebrovascular episodes is between 20 and 40 years (Yanagawa et al, 2002). Associated features are alopecia, spondylosis deformans, and psychiatric disorders (Fukutake, 1999; Yanagawa et al, 2002).
Diagnosis: Magnetic resonance imaging shows aspecific white matter signal abnormalities and lacunar lesions. The causative gene is unknown and the diagnosis can be suggested from clinical presentation, pattern of inheritance, MRI appearance, and autopsy confirmation of arteriosclerosis without GOM (Arima et al, 2003; Fukutake, 1999).
Hereditary Endotheliopathy with Retinopathy, Nephropathy, and Stroke
Hereditary endotheliopathy with retinopathy, nephropathy, and stroke is a rare autosomal-dominant, multiinfarct syndrome with generalized vasculopathy.
Epidemiology: It is exceptionally rare and only few tens of patients have been reported (Jen et al, 1997).
Genetic and Pathological Aspects: Genetic analysis has mapped the disease locus to chromosome 3p21.1–p21.3 (Cohn et al, 2005; Ophoff et al, 2001), but the pathologic basis remains unclear. Ultrastructural studies revealed alterations in arterioles and capillaries consisting in multilayered basement membrane in the brain and in other tissues such as kidney and skin biopsies (Jen et al, 1997).
Clinical Phenotype: The clinical symptoms are the consequence of the diffuse endotheliopathy; the onset is usually in the third and fourth decade of life (Jen et al, 1997). Initial manifestation is typically a progressive retinopathy with visual loss, followed by psychiatric symptoms and neurologic manifestations, such as stroke, migraine-like headache, and dementia. In addition, several patients had a renal dysfunction with azotemia, proteinuria, or hematuria (Jen et al, 1997; Cohn et al, 2005).
Diagnosis: Electron microscopy of skin biopsy shows alterations in the arterioles and capillaries consisting of multilayered basement membranes. Magnetic resonance imaging reveals contrast-enhancing subcortical lesions with surrounding edema, especially in the frontoparietal region (Jen et al, 1997). However, the diagnosis is confirmed by genetic analysis.
Disorders affecting both small and large arteries
Fabry Disease
Fabry disease (FD) is an X-linked recessive lysosomal storage disorder, with multisystemic involvement, resulting from the deficient activity of the enzyme α-galactosidase A (α-Gal A) (Brady, 1967; Brady et al, 1967).
Epidemiology: The classic phenotype of FD has an estimated incidence of about one in 40,000 to 117,000 male live births (Desnick et al, 2001; Meikle et al, 1999). However, the diagnosis of FD is often missed or delayed, particularly in the absence of the characteristic manifestations of the classic phenotype. Recently, a screening of 37,104 consecutive newborn males found that 12 neonates had deficient α-Gal A activities and specific mutations, revealing a surprisingly high FD incidence of one in ≈3, 100 males. Of the 12 neonates with α-Gal A deficiency, 11 had mutations predicting the later-onset phenotype for an 11:1 ratio of later-onset/classic phenotypes (Spada et al, 2006).
Genetic and Pathological Aspects: Fabry disease results from mutations of the α-Gal A gene at Xq22. More than 200 mutations distributed over the entire α-Gal A gene have been reported, and they include all types of gene defects (Pastores and Lien, 2002; Schafer et al, 2005). Most of the mutations have been found in single families (Garzuly et al, 2005; Sorensen, 2002). The deficit of α-Gal A enzyme is responsible for the accumulation of glycosphingo-lipids, particularly globotriaosylceramide, within the heart, kidney, brain, peripheral nerves, eyes, skin, and vascular tissues. Globotriaosylceramide is stored particularly in the vascular smooth muscle and endothelial cells with cell dysfunction and development of tissue ischemia and infarction. The deposition of globotriaosylceramide was documented within the cerebral vascular wall causing narrowing of perforating small arterioles and dolichoectasia of the large basilar and vertebral arteries (Garzuly et al, 2005). Garzuly et al (2005) described a family with megadolichobasilar and thrombosis occurring in five men and a woman caused by a mutation (c.47T-> C missense mutation) in α-Gal A gene.
Clinical Phenotype: Cerebrovascular disorders have an incidence of about 40% in young homozygous male individuals, and the average age at the onset of cerebrovascular symptoms is 33.8 years (Kolodny and Pastores, 2002). Ischemic episodes occur due to the damage of small and large blood vessels, but also due to cardiogenic embolism resulting from myocardial abnormalities (Kolodny and Pastores, 2002). Stroke or TIA occur mainly in the posterior circulation and infrequently in the anterior one (Crutchfield et al, 1998; Mitsias and Levine, 1996), but the reason of this distribution is unclear. Later-onset variants of FD, with residual α-Gal A activity due to missense mutations, have been identified in individuals who lack the early FD manifestations (i.e., angiokeratoma, acroparesthesias, hypohidrosis, and corneal/lenticular abnormalities) (Nakao et al, 1995, 2003; von Scheidt et al, 1991). Recently, a new phenotype of FD, characterized by unexplained strokes and without the early classic manifestations, has been reported. The patients with this type of FD presented cryptogenetic stroke, especially in the vertebrobasilar artery system, and proteinuria (Rolfs et al, 2005). Mitsias and Levine (1996) also described cerebral hemorrhages due to aneurysmal deformity of blood vessels. Another neurological manifestation is a small-fiber polyneuropathy characterized by painful acroparesthesias, hypohidrosis, and autonomic dysfunctions (Kolodny and Pastores, 2002). Instead, multisystemic manifestations include characteristic skin lesions (angiokeratomas), a corneal opacity that does not affect vision (cornea verticillata), gastrointestinal disorders, and other organ diseases (kidney and heart). Renal involvement is frequently seen in affected males, initially with proteinuria and ultimately with severe renal failure (Grunfeld et al, 2001). Cardiovascular complications include left ventricular hypertrophy, valvular structural abnormalities, conductions disturbances, and coronary artery disease (Linhart et al, 2001). Manifestations of the disease have also been reported in heterozygote females, ranging from most being asymptomatic or having mild manifestations to rare cases having a severe disease as in affected males. This has a possible explanation in the skewed X-chromosome inactivation (Deegan et al, 2006; MacDermot et al, 2001). In embryonic development, one of the two X chromosomes in each somatic cell becomes inactivated, which results in patchy and variable expression of the detective gene. Mehta et al (2004) noted that, in a cohort of 366 patients with FD (201 males and 165 females), the frequency of stroke or TIA was higher in women than in men, although in the females the cerebrovascular events occurred later in life.
Diagnosis
Neuroimaging: Magnetic resonance imaging can show arterial dolichoectasia, extensive periventricular white matter signal intensity abnormalities (leukoaraiosis), and large or small vessel infarcts. The MRI findings are not specific except for hyperintensity in the pulvinar region on T1-weighted images. This is a distinctive characteristic but present in only 24% of patients (Moore et al, 2003).
Skin biopsy: Skin biopsy can show typical lipid inclusions within the cytoplasm of various dermic cells and also adipocytes of normally appearing skin (Le Charpentier et al, 1981). However, biochemical analysis is required to confirm the clinicopathological suspicion (Linthorst et al, 2004).
Biochemical studies: In affected males, the diagnosis is confirmed by measuring the α-Gal A activity in plasma or in peripheral leukocytes. In females, α-Gal A activity may be normal; therefore, the diagnosis should be confirmed by the detection of gene mutations (Desnick et al, 2003).
Genetic analysis: The diagnosis is confirmed by the detection of mutations of the α-Gal A gene at Xq22.
Treatment: The past treatment of FD consisted of symptomatic and palliative managements of pain and organ failure. Recent randomized-controlled trials showed that recombinant α-Gal A enzyme improves and reverses signs and symptoms of FD. In particular, substitutive therapy decreases the level of neuropathic pain, improves renal dysfunction, and reduces electrocardiogram abnormalities (Eng et al, 2001; Schiffmann et al, 2001), but any beneficial effect on the cerebrovascular manifestations is difficult to show (Mehta and Ginsberg, 2005).
Pseudoxanthoma Elasticum
Pseudoxanthoma elasticum (PXE) is a heritable connective disorder characterized by skin, eyes, and cardiovascular complications.
Epidemiology: The prevalence was estimated to be approximately one per 70,000 to 100,000 (Schievink et al, 1994; Struk et al, 1997), but it might be higher for the variable expression and penetrance of the disease (Trip et al, 2002) and for the lack of knowledge about PXE by physicians.
Genetic and Pathological Aspects: Pseudoxanthoma elasticum is related to mutations in the ABCC6 (ATP-BINDING CASSETTE, SUBFAMILY C, and MEMBER 6) gene on chromosome 16p13,1 (Bergen et al, 2000; Struk et al, 1997). ABCC6 consists of 31 exons in which almost 90 different mutations have been reported (Chassaing et al, 2005). Pseudoxanthoma elasticum was found not only in sporadic patients but also in families with autosomal-recessive and -dominant inheritances. However, no specific dominant mutation has been described, and autosomal-dominant inheritance remains uncertain (Plomp et al, 2004). ABCC6 encodes the multidrug resistance-associated protein 6, an ATP-dependent transmembrane transporter protein (Ringpfeil et al, 2000). ABCC6 is predominantly expressed in the liver and kidney, but low expression levels have been detected in other affected tissues. The exact physiological function of ABCC6 is unknown, and the correlation between genotype and phenotype has not been established. The histopathological feature is the presence of abnormal, mineralized, and fragmented elastic fibers in skin, eyes, and arterial blood vessels (elastorrhexia) (Chassaing et al, 2005).
Clinical Phenotype: Considerable clinical intra- and interfamilial variability was observed (Gheduzzi et al, 2004). Three organ systems (cardiovascular system, skin, and eyes) are involved in some patients, whereas in others, there is a limited involvement of just one of these organs (Sherer et al, 2001). Abnormalities in elastic fibers of arterial walls are the cause of vascular manifestations, including hypertension, angina pectoris, intermittent claudication, restrictive cardiomyopathy, mitral valve prolapse, or stenosis. Also, bleeding complications occur especially in the gastrointestinal tract and less commonly in the cerebrovascular system, uterus, and urinary tract (Chassaing et al, 2005; Heaton and Wilson, 1986). Ischemic stroke is caused by large artery disease or, less frequently, by small-vessel occlusive disease. Moreover, hypertension, very common in patients with PXE, favors cerebrovascular episodes (Chassaing et al, 2005; Pavlovic et al, 2005). In patients with PXE, symptomatic intracranial aneurysms were also reported but an association between the two diseases is unlikely (van den Berg et al, 2000). The skin lesions are yellowish papules and plaques predominantly located on the neck, popliteal fossae, antecubital, inguinal, axillae, and peri-umbilical areas. Ocular signs are a mottled hyperpigmentation of the retina (peau d'orange appearance) and angioid streaks, which result from degeneration of the Bruch's membrane, the elastic-rich layer of the retina. The damage of chorioid and retinal vessels can be complicated by aberrant neovascularization and retinal hemorrhages, which lead to visual impairment (Chassaing et al, 2005; Hu et al, 2003).
Diagnosis
Skin biopsy: The presence of pleomorphic, fragmented, and calcified elastic fibers at skin biopsy can suggest the diagnosis, and abnormal elastic fibers can also be found in apparently normal skin (Lebwohl et al, 1987). However, the skin histology is not pathognomonic for PXE because other diseases (e.g., penicillamine intoxications) are histologically difficult to distinguish (Hu et al, 2003).
Genetic analysis: The clinical and histopathological diagnosis can be difficult and the identification of gene defect is necessary (Christen-Zach et al, 2006).
Treatment: Individuals with PXE should adopt prophylactic measures and lifestyle rules to reduce the risks of complications. Contact sports should be avoided, and the use of platelet inhibitors and anticoagulants is controversial for the high incidence of bleeding (Laube and Moss, 2005; Pavlovic et al, 2005; van den Berg et al, 2000). Moreover, nutritional restriction of calcium has been suggested because a correlation of high calcium intake and severity of PXE has been observed (Laube and Moss, 2005).
Neurofibromatosis Type 1
Neurofibromatosis type 1 (NF1), also called von Recklinghausen disease or peripheral neurofibromatosis, is a multisystem disorder involving tissue of mesodermal and ectodermal origin (Schievink et al, 1994).
Epidemiology: The incidence was estimated to be approximately 1/3500 individuals (Friedman, 1999).
Genetic and Pathological Aspects: Neurofibromatosis type 1 is an autosomal-dominant disorder with complete penetrance but variable expression. It results from mutations in the NF1 gene on chromosome 17q11.2. The gene spans over 350 kb of genomic DNA and encodes an mRNA of 11 to 13 kb containing at least 59 exons (Shen et al, 1996). The NF1 gene product termed ‘neurofibromin,’ is a large tumor suppressor protein. Loss of neurofibromin produces increased mitogenic signaling and leads to cellular proliferation or differentiation (Hamilton and Friedman, 2000; Weiss et al, 1999). Neurofibromin is expressed principally in neurons, Schwann cells, oligodendrocytes, astrocytes, leukocytes and also in blood vessels, muscle, and skin (Daston et al, 1992; Norton et al, 1995; Ward and Gutmann, 2005). Histological features of affected blood vessels are an abnormal proliferation of spindle cells and of intima with fibrous thickening and mesodermal dysplasia or fibromuscular hyperplasia. Micronodular formations of smooth muscle aggregates appear on the walls of vessels. The vascular abnormalities involve both small and large vessels and lead to stenosis, occlusion, or rupture of the blood vessels (Hamilton and Friedman, 2000).
Clinical Phenotype: Organ systems involved in NF1 include skin (neurofibromas, café-au-lait spots, and freckling), eyes (Lisch nodules), peripheral nervous system (neurofibromas and schwannomas), meninges (dural ectasia), skeleton (scoliosis, dysplasia), endocrine system (pheochromocytomas), and vascular system (Schievink et al, 1994). Stroke in patients with NF1 is rare. On examining 158 NF1 patients, Creange et al (1999) observed neurological manifestations in 55% of these, and only one had a history of stroke. However, the vascular manifestations are underestimated among NF1 patients, because the affected individuals are often asymptomatic. Vascular lesions identified in NF1 are stenosis, occlusion, pseudoaneurysm, aneurysm, ectasia, rupture of large and medium-sized arterial, arteriovenous malformation, and fistulae (Norton et al, 1995; Schievink et al, 1994). The internal carotid and the middle or anterior cerebral arteries are more frequently involved than the posterior circulation (Rosser et al, 2005). The occlusive lesions can lead to development of collateral circulation, resulting in the ‘moyamoya’ appearance on angiographic evaluation (Friedman et al, 2002). Other neurological manifestations are epilepsy and headache (Creange et al, 1999).
Diagnosis
Clinical criteria: Diagnosis of neurofibromatosis is based on the presence of two or more of the following features: (1) six or more café-au-lait spots (> 1.5 cm in postpubertal individuals and > 0.5 cm in prepubertal individuals); (2) two or more neurofibromas of any type or one plexiform neurofibroma; (3) freckling in the auxillary or inguinal regions; (4) optic pathway glioma; (5) dysplasia of the sphenoid bone or of the long bones; (6) first-degree relative with NF1 (Ward and Gutmann, 2005).
Genetic analysis: The presence of mutations in NF1 gene confirms the clinical diagnosis.
Treatment: No medical treatment is available to prevent the characteristic lesions, and surgery or chemotherapy are currently the only treatment option for most of the lesions in NF1 (Tonsgard, 2006). There is no specific therapy for the cerebrovascular complications.
Large artery diseases
Familial Moyamoya Disease
Moyamoya disease is a nonatherosclerotic, noninflammatory, and nonamyloid cerebrovascular disorder characterized by angiographic findings of bilateral progressive stenosis of the terminal portion of the internal carotid artery with development of fragile collateral circulation, the ‘moyamoya’ vessels (Suzuki and Takaku, 1969).
Epidemiology: The prevalence of Moyamoya disease is higher in the Japanese (≥3 per 100,000 persons) (Suzuki and Takaku, 1969) and oriental populations, even if all races could be affected (Battistella and Carollo, 1997; Borota et al, 1997; Numaguchi et al, 1997; Yonekawa et al, 1997).
Genetic and Pathological Aspects: The etiology of this disease is unknown, but its prevalence in the Japanese, the familial cases, and the high concordance in monozygotic twins suggest a hereditary basis. Osawa et al (1995) concluded that it is inherited most probably in a polygenic mode or in an autosomal-dominant manner with a low penetrance. In 1999, the gene responsible for familial Moyamoya disease was mapped on chromosome 3p24.2–p26 (Ikeda et al, 1999), and in 2000, another locus was mapped on chromosome 17q25 (Yamauchi et al, 2000).
Histologically, there is a stenosis or an obstruction of vessels by noninflammatory, nonarteriosclerotic thickening of the intima with layered elastic laminae and few lipid deposits. These findings predominantly affect the intracranial vessels, but the vascular changes also involve extracranial vessels such as renal, coronary, pancreatic, and pulmonary arteries (Ikeda, 1991).
Clinical Phenotype: Cerebral ischemia predominates in children and cerebral hemorrhage in adults. Stenosis or obstruction of the vessels causes brain ischemia before 10 years of age, and progressive narrowing of carotids results in development of collateral fragile new vessels whose rupture causes hemorrhage (subarachnoid, intraparenchymal, or intraventricular), predominantly after 30 years of age (Bruno et al, 1988; Fukui, 1997; Han et al, 2000; Ikezaki et al, 1997a, b).
Diagnosis: Moyamoya disease is defined as bilateral stenosis or occlusion of the internal carotid artery of unknown origin, and cerebral angiography is essential for diagnosis. However, unilateral involvement and posterior cerebral artery stenosis/occlusion were also reported (Houkin et al, 1996; Ikezaki et al, 1997a, b; Kuroda et al, 2002). The diagnostic criteria (Fukui, 1997) classify cases with bilateral and unilateral involvement of internal carotid artery, respectively, as definite and probable Moyamoya disease. Because the etiology of this disease is unknown, similar lesions associated with other conditions (e.g., head trauma, autoimmune disease, and Recklinghausen's disease) should be excluded from the diagnosis.
Treatment: The best therapy for this disease remains unknown. Patients can be treated with conservative therapy, such as antihypertensive and fibrinolytic agents, or with surgical revascularization. Different surgical revascularization procedures have been used in patients with ischemia, but randomized controlled trials that compared surgical revascularization techniques with medical therapy or with each other have not been done (Han et al, 2000).
Arterial dissection
Ehlers-Danlos Syndrome Type IV
The Ehlers-Danlos syndromes (EDS) are a heterogeneous group of heritable disorders of connective tissue. Eleven subtypes have been recognized on the basis of clinical and genetic differences, although a recent revision of EDS classification has simplified it from 11 to six distinct varieties: classical, hypermobility, vascular, kyphoscoliosis, arthrochalasia, and dermatosparaxis types (Beighton et al, 1998). The vascular EDS syndrome is known as type IV.
Epidemiology: Estimated prevalence of EDS is one in every 10,000 to 20,000 births. The vascular type (EDS type IV) is rare, accounting for < 4% of EDS patients (Germain, 2002; Oderich et al, 2005).
Genetic and Pathological Aspects: The Ehlers-Danlos syndrome type IV is an autosomal-dominant inherited disorder, resulting from mutations in the COL3A1 gene, located in the long arm of chromosome 2 at 2q24.3–q31 (Germain, 2002). COL3A1 includes approximately 52 exons, and more than 200 different mutations have been found (nonsense or missense mutations, incorrect splicing with exon skipping, or deletion) (De Paepe and Malfait, 2004; Germain, 2002; Pepin et al, 2000; Superti-Furga et al, 1988). The COL3A1 gene encodes the proα1(III) chain of type III collagen, a member of the fibrillar collagen family, localized mostly in blood vessels and skin (De Paepe and Malfait, 2004). The blood vessels of affected patients have an abnormally low intima media thickness inducing vessel fragility and predisposition to dissection and rupture (Boutouyrie et al, 2004).
Clinical Phenotype: Vascular fragility involves large and small vessels in every site in the body, and it is the principle responsible for clinical manifestations. The most common central nervous system events are fistulaes involving the carotid artery and cavernous sinus, carotid-artery dissections, aneurysms, and arterial ruptures (Oderich et al, 2005; Pepin et al, 2000). Stroke is uncommon, but early diagnosis is particularly important to weigh the pros and cons of further examinations and procedures with high risk of complications such as cerebral angiography. Patients with vascular EDS often have a distinctive facial appearance, including prominent eyes, a thin, pinched nose, small lips, hollow cheeks, and lobeless ears (Germain, 2002). Other clinical features are dermatological manifestations (easy bruising and translucent, thin, and hyperextensible skin), gastrointestinal perforation, obstetrical complications, and rarely joint hypermobility, usually limited to the small joints of the hands (Germain, 2002).
Diagnosis
Diagnostic criteria: The clinical diagnosis is based on the findings of at least two of four diagnostic criteria that are thin, translucent skin, arterial, intestinal or uterine rupture, extensive bruising, and characteristic facial appearance (Beighton et al, 1998).
Skin biopsy: The clinical diagnosis is confirmed by biochemical studies that identify quantitative (cultured skin fibroblast synthesizes reduced type III procollagen) and qualitative (abnormal migration at electrophoresis) defects of type III collagen (Beighton et al, 1998).
Genetic analysis: The demonstration of a mutation in the COL3A1 gene confirms the diagnosis (Beighton et al, 1998).
Treatment: There are only symptomatic treatments, genetic counseling and precautionary measures such as avoiding lifestyles with increased potential for trauma (contact sports), invasive vascular procedures and drugs that interfere with coagulation and platelet function (Oderich et al, 2005). Unless absolutely essential for adequate treatment, noninvasive investigations should be preferred to angiography, because it has a high risk of complications (Oderich et al, 2005).
Marfan Syndrome
Marfan syndrome (MFS) is an autosomal-dominant inherited disorder of connective tissue.
Epidemiology: It has a prevalence of two to three in 10,000 individuals (Pyeritz, 2000).
Genetic and pathological aspects: This disease is caused by mutations on fibrillin-1 (FBN1) gene in more than 90% of cases (Loeys et al, 2004) and on transforming growth factor-β receptor 2 (TGFβR2) gene in a few cases (Mizuguchi et al, 2004). In 2004, TGFβR2 at 3p24.1 was identified as the Marfan syndrome type II gene, and this gave the first genetic evidence of a link between abnormal TGF-β signaling and a human connective tissue disorder (Boileau et al, 2005). FBN1 gene on chromosome 15q21 contains 65 coding exons and more than 500 mutations have been identified without predilection for any given region. The genotype—phenotype correlation is not clear and identical mutations have been associated with different forms of MFS (Boileau et al, 2005). FBN1 gene encodes a glycoprotein called fibrillin-1 that is an important component of the microfibrillary system and has a wide tissue distribution. Defects in fibrillin are associated with damage of elastic fibers and may predispose to aneurysm formation and arterial dissection (Robinson et al, 2002).
Clinical Phenotype: The MFS has highly variable clinical manifestations with prominent involvement of skeletal, ocular, and cardiovascular systems. Intracranial aneurysms and spontaneous dissection of carotid and vertebral arteries, causing stroke, have been reported in patients with MFS syndrome (Schievink et al, 1994). These complications are estimated to occur in 10 to 20% of patients (Schievink et al, 1994). Also a high risk of cardioembolic stroke was estimated in these patients. In a retrospective hospital-based study of patients with MFS, seen in an 8-year period, the incidence of neurovascular disorders was 3.5%; ischemic events were 83%; hemorrhagic events 17%. Prosthetic heart valves and atrial fibrillation embolism were the most common cause of neurovascular disorders in this study (Wityk et al, 2002). Wityk et al, noting that most strokes are cardioembolic, cast doubt on the association between spontaneous, isolated internal carotid artery dissection and MFS. Other two studies raised a similar question of whether there is truly an association of intracranial aneurysm with MFS (Conway et al, 1999; van den Berg et al, 1996). The skeletal manifestations include tall stature, pectus excavatum, or carinatum, high-arched palate, arachnodactyly, laxity of ligaments with scoliosis, or joint hyperextensibility (Pyeritz, 2000). Other clinical features are ocular manifestations (strabismus, amblyopia, ectopia lentis, and cataract), cardiovascular manifestations (dissection or dilatation of ascending aorta, mitral valve prolapse, and cardiac electrical disturbances), and dural ectasia (Pyeritz, 2000).
Diagnosis
Diagnostic criteria: Because at present, it is not possible to find a mutation in about 35% of the patients, the diagnosis of Marfan syndrome is primarily based on clinical criteria (Loeys et al, 2001).
Diagnostic criteria established by Beighton et al and revised by De Paepe et al (1996) are based on a combination of major and minor clinical manifestations in different organ systems. Major criteria (presence of skeletal manifestations, ectopia lentis, aortic dilatation/dissection, lumbosacral dural ectasia, and a positive family history) have a high diagnostic specificity because they are infrequent in other conditions and in the general population. Minor criteria, such as joint hypermobility, mitral valve prolapse, and spontaneous pneumothorax, have a low specificity because they are frequent in the general population. The diagnosis in an index case, without a genetic/family history, requires major criteria in at least two different organ systems and involvement of a third organ system (De Paepe et al, 1996).
Genetic analysis: Mutations in FBN1 and TGFβR2 gene confirm the diagnosis.
Treatment: Patients with MFS require a multidisciplinary management that includes a geneticist, an ophthalmologist, a cardiologist, and an orthopedist. About cardiovascular complications, patients should avoid contact sport or competitive sport and isometric exercise to reduce the risk of acute aortic dissection. Medical treatment with b-adrenergic blockade seems to protect the aorta from dilatation and dissection; moreover, it is recommended that regular imaging is performed to detect and quantify progression of aorta dilatation, and in some cases, surgery may become necessary (Judge et al, 2005; Pyeritz, 2000). All people with MFS should use routine antibiotic prophylaxis before dental or other procedures that carry a high risk of introducing bacteria in the blood steam (Pyeritz, 2000).
Discussion
This review is intended for physicians who will be seeing stroke patients and who have a specific interest in cerebrovascular disease. To make clearer the description of monogenic disorders, we listed the diseases according to stroke mechanism (small vessel, large vessel, a combination of both and arterial dissection; Table 1). For each disease, our paper overviews concisely the most important clinical manifestations and focuses extensively on current understanding of cerebrovascular aspects (the main characteristics of each disease are reviewed in Table 2). Young age at stroke onset (< 50 years), absence of conventional stroke risk factors, and a family stroke history are often perceived as evidence of an underlying monogenic disorder (Hassan et al, 2002), but although these findings could support the diagnostic suspicion, they do not necessarily mean a single-gene disorder. The causes of stroke among young adults are diverse (spontaneous cervical arterial dissection, patent foramen ovale, IgG anticardiolipin antibodies, and so on), and a positive family history in patients younger than 65 years was frequently seen in an unselected group of patients with stroke or TIA (Hassan et al, 2002). Since stroke may be only a manifestation of a multisystemic monogenic disease, there are several signs and symptoms that should be investigated to support a suspicion of a single-gene disorder (Table 2). The presence of migraine, cognitive impairment, or psychiatric features may suggest a small vessel disease (CADASIL, cerebral autosomal-recessive arteriopathy with subcortical infarcts and leukoencephalopathy, and hereditary endotheliopathy with retinopathy, nephropathy, and stroke), skeletal abnormalities, and eye involvement may underlie a connective disorder (pseudoxanthoma elasticum, Ehlers-Danlos syndrome type IV, and Marfan syndrome), and the failure of different tissues may prompt a multisystem disease (FD and neurofibromatosis type 1). A further useful finding, which could address a particular monogenic disorder, is the identification of a specific stroke subtype. In the majority of cases, a specific single-gene disorder is related to a specific stroke phenotype, although some disorders, such as FD, predispose to stroke by more than one mechanism. Vascular and parenchymal lesions seen with MRI and angiography could be a good support to the clinical diagnosis. In patients with CADASIL or FD, the brain MRI can show specific lesion patterns, and in Moyamoya disease, angiography findings could be diagnostic. Another rapid and noninvasive investigation is skin biopsy, which permits the diagnosis to be addressed in a number of cases (CADASIL, hereditary endotheliopathy with retinopathy, nephropathy, and stroke, Fabry, PXE, and EDS). Therefore the presence of specific clinical systemic features, in addition to young age at stroke onset, the lack of vascular risk factors, and a positive family stroke history could suggest a single-gene disorder. Thus specific diagnostic tools can aid the diagnosis, but genetic screening is nearly always necessary to confirm the diagnostic suspicion.
Monogenic causes of ischemic stroke
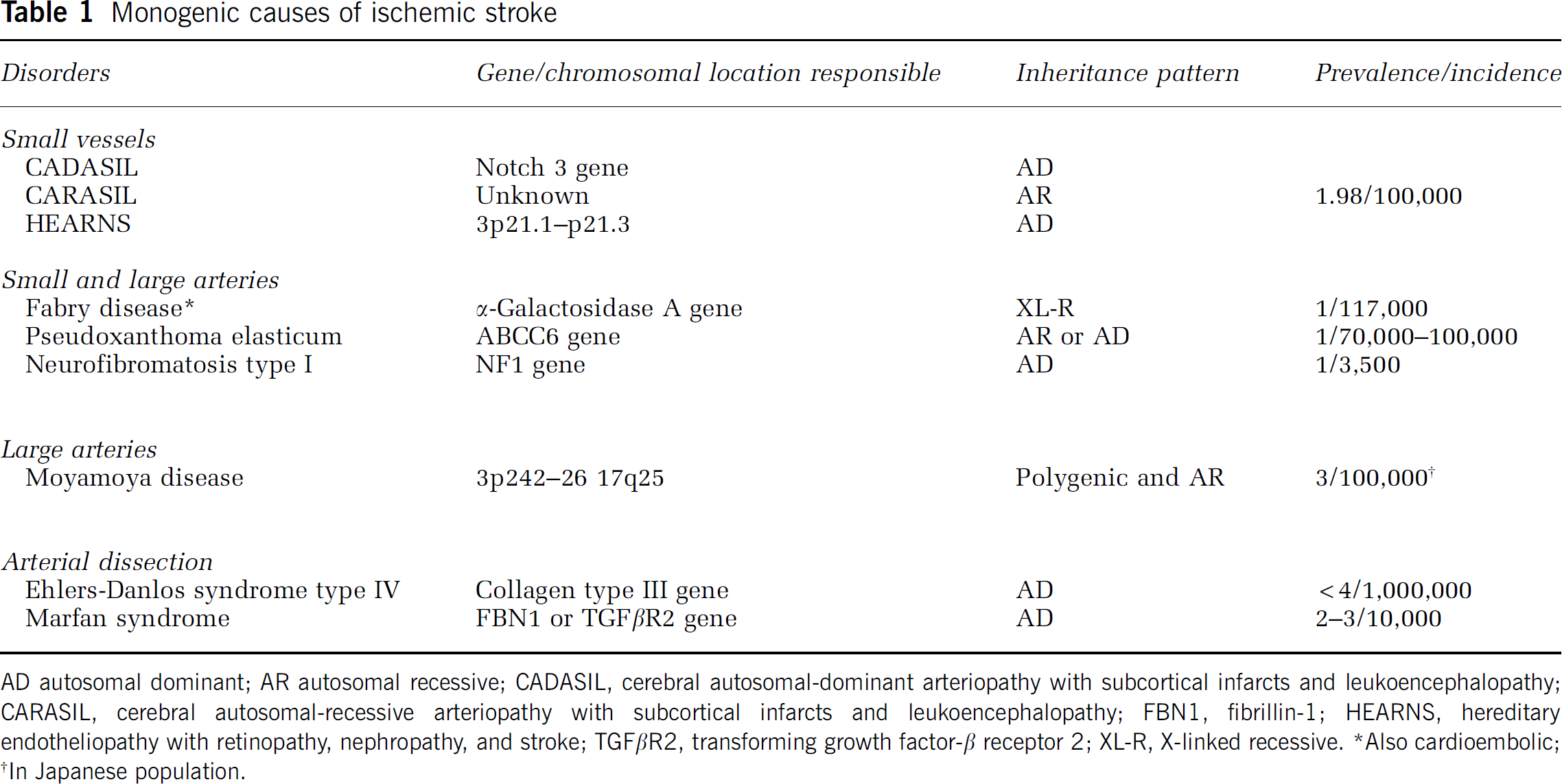
AD autosomal dominant; AR autosomal recessive; CADASIL, cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy; CARASIL, cerebral autosomal-recessive arteriopathy with subcortical infarcts and leukoencephalopathy; FBN1, fibrillin-1; HEARNS, hereditary endotheliopathy with retinopathy, nephropathy, and stroke; TGFβR2, transforming growth factor-β receptor 2; XL-R, X-linked recessive.
Also cardioembolic
In Japanese population.
Indications for the diagnosis of monogenic ischemic stroke
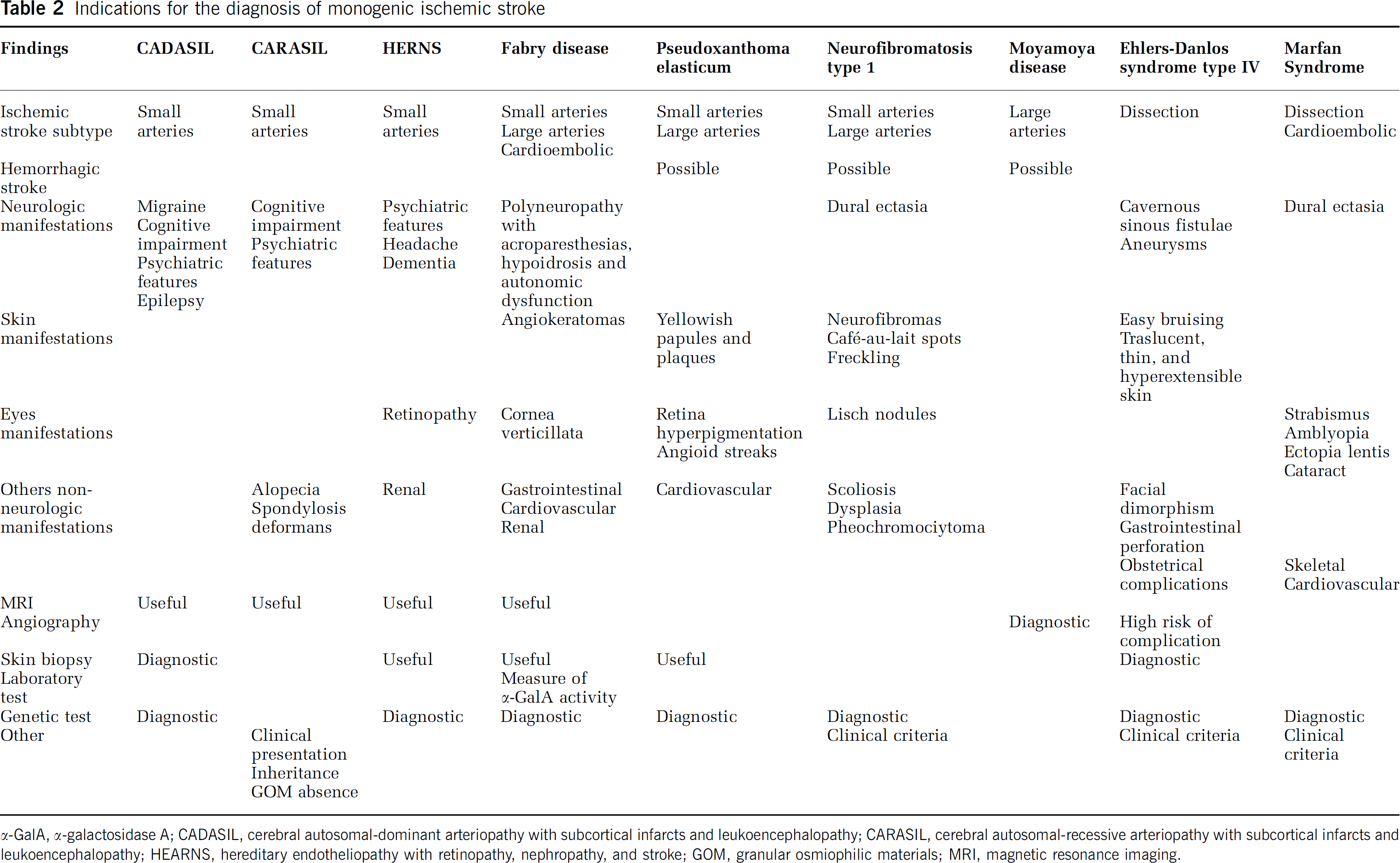
α-GaIA, α-galactosidase A; CADASIL, cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy; CARASIL, cerebral autosomal-recessive arteriopathy with subcortical infarcts and leukoencephalopathy; HEARNS, hereditary endotheliopathy with retinopathy, nephropathy, and stroke; GOM, granular osmiophilic materials; MRI, magnetic resonance imaging.