
Abstract
Dietary therapies represent a potentially valuable adjunct to other epilepsy treatments, such as anticonvulsant medications, epilepsy surgery, and vagus nerve stimulation. Although the ketogenic diet (high fat, adequate protein, low carbohydrate) is the most well-established dietary therapy for epilepsy, other possible approaches include the Atkins diet (high fat, high protein, low carbohydrate), a diet enriched in polyunsaturated fatty acids, or overall restriction of calorie intake. This review discusses the current clinical status of each of these dietary approaches and suggests possible mechanisms by which they might suppress neuronal hyperexcitability and seizures.
The prospect that epilepsy might be controlled, at least partially, by nutritional modification is radical but highly appealing. The ketogenic diet (KD) is certainly the best-known dietary approach to epilepsy treatment, but it is not the only one (1). This review updates the clinical and basic mechanistic information available about the KD, as well as other dietary therapies, including the Atkins diet, calorie restriction, and a diet enriched in polyunsaturated fatty acids. Emphasis is placed on possible mechanisms by which each diet might enhance seizure control. Clinical details can be found in recent reviews (2–5).
Examples of Dietary Approaches to Epilepsy Treatment
Ketogenic Diet
By now, the KD is well known to the epilepsy community. It was initially devised in 1921 to mimic the anticonvulsant effects of fasting, which were known to suppress seizures (6). The formulation and administration of the KD have varied little since the 1920s. The KD was used extensively until anticonvulsant drugs (AEDs) became available, beginning in the 1930s. The use of the KD continued through the decades, but mainly as a “last resort.” In the 1990s, KD use underwent a resurgence. It now holds an important place in the standard armamentarium of epilepsy treatments but is still used primarily in children (and increasingly in adults) with seizures refractory to standard AEDs.
The clinical efficacy of the KD has been verified in numerous studies, both in the United States and internationally (7–13). The success rate of the KD in controlling refractory seizures is at least as good as, and often better than that of the “new” AEDs (10). In general, at least half of all patients treated with the KD will exhibit a 50% or greater reduction of seizure frequency. Any seizure type may respond to the diet (14), but some generalized seizure types [e.g.,myoclonic, atonic, generalized tonic–clonic, and even infantile spasms (15)] may be reduced preferentially. The KD is effective in people of all ages, although it may be maximally effective in the toddler and school-age child (8,16,17). Interestingly, the success rate of the KD in recent years is similar to that during the early decades of its use (18). Perhaps most important, intriguing recent data indicate that a KD sometimes can be discontinued without concomitant loss of seizure control (2,11). This observation suggests that the KD might be both anticonvulsant and antiepileptogenic.
Unfortunately, after nearly a century of use, the mechanism of action of the KD remains unclear. Many clinical issues remain unresolved regarding the KD and its use, including the necessity of starting the diet with a period of fasting (19), the need for fluid restriction, the seizure types most benefited, the patients most amendable to it use, and its long-term benefits and side effects (20). Few animal studies of the KD were performed prior to 1996 [reviewed in (21)]. In recent years, several laboratories have embarked on efforts to determine the biochemical and physiologic basis of how the KD works. Any explanation of the KD mechanism of action must take into account certain clinical observations and known biochemical alterations resulting from ingestion of the KD, as described in Table 1.
Animal models of the ketogenic diet: observations and clinical correlates

Adapted from Stafstrom CE, et al., 2003
KD, ketogenic diet; MCTs, medium-chain triglycerides; PUFA, polyunsaturated fatty acid.
When switching from a diet high in carbohydrates to one high in fats and with stringent carbohydrate restriction, the body uses fats as the primary energy source. Fat breakdown in the liver creates ketone bodies (β-hydroxybutyrate, acetoacetate, and acetone), which circulate to the brain and are taken up into cerebral tissue via specific monocarboxylate transporters (22). In neuronal mitochondria, ketones are metabolized to adenosine triphosphate (ATP) via the tricarboxylic acid cycle and oxidative phosphorylation. The challenge has been to understand how this energy shift results in an anticonvulsant effect. Obviously, multiple sites exist in the relevant biochemical pathways at which seizure suppression could be facilitated, and the mechanism (or more likely, mechanisms) by which the KD exerts an anticonvulsant effect likely involves the combination of the altered energy homeostasis and regulation of neuronal and synaptic excitability.
Early theories of KD mechanism focused on the involvement of ketone bodies. This hypothesis would make sense, because serum ketone concentration increases markedly in subjects on the KD, and urinary ketones are used clinically as the marker of ketosis. However, several clinical and experimental studies have shown that the relation between serum or urinary ketones and seizure control is imprecise, at best (23–26). Direct effects of ketones on neurophysiologic parameters, excitatory and inhibitory neurotransmission, and ictal activity in vitro have been essentially negative (27,28), although the inclusion of glucose in the bathing medium of the brain slices in those experiments could have counteracted the influence of the ketones. Indeed, recent experiments have shown that, in vivo, rats on the KD or a calorie-restricted normal diet exhibited increased paired-pulse inhibition in the hippocampal dentate gyrus and increased resistance to maximal dentate activation (a form of seizure activity) (29). It is possible that the volatile ketone body acetone could exert an anticonvulsant effect (30–33), and a reliable measurement system of breath acetone concentration has been developed (34). At present, we may conclude that some, as yet undetermined, “threshold level” of ketosis is required and must be maintained for the KD to be maximally effective. Whether seizure control correlates with ketone levels remains unclear; the concentration of ketones at the synapse is unknown but may be a critical parameter.
The alteration in cerebral energetics induced by the KD favors an increase in “energy charge,” that is, a relative increase in the ATP/adenosine diphosphate (ADP) ratio resulting from metabolic alterations of the enzymes involved in glycolysis and the tricarboxylic acid cycle (35,36). The greater availability of energy may reduce cellular excitability, by enhancing energy available for cellular processes, such as membrane pumps and transporters, which enhance hyperpolarization (37). The energy-charge hypothesis has received some support from recent studies in fasting humans, by using [31P]-spectroscopic imaging, in which ketones transported from the blood to the brain are used by neurons (38).
It is possible that γ-aminobutyric acid (GABA), the main inhibitory neurotransmitter in the brain, plays a role in how the KD decreases neuronal excitability. Enhancement of GABA function by the KD could occur through pathways of ketone body metabolism (GABA shunt), whereby glial conversion of glutamate to glutamine (a GABA precursor) is enhanced by ketosis (39). Ketones also mimic GABA structurally and could play a direct inhibitory role on cellular excitability by stimulating GABA receptors or enhancing their action (40,41). KD treatment (and calorie restriction) increases the expression of glutamic acid decarboxylase (GAD65 and GAD67) isoforms, providing a mechanism by which GABA synthesis might be increased in a patient on the KD (42). In a study using a variety of seizure-induction methods, the most consistent protection with the KD was found when seizures were induced by blocking GABA receptors with picrotoxin, bicuculline, or (γ-butyrolactone (43).
An alternative mechanism for KD action would implicate the type or quantity of fat in the diet. Typically, a KD includes mostly saturated fats, usually in the form of heavy cream or butter. Fats in the classic KD consist of a mixture of animal-and plant-derived fats; fatty acids of various chain lengths are likely to be included, but no attempt is made to specify fat type or chain length in the diet formulation. An exception is the medium-chain triglyceride (MCT) diet, in which oils containing certain chain lengths are the main source of fat (44). Early clinical studies showed a correlation between plasma lipids and seizure control in children with epilepsy treated with the KD (45). Plasma lipid levels peaked as KD-induced seizure control became maximal. In experimental studies, young rats fed a variety of different fat types developed seizure resistance independent of the level of ketosis (26). The authors concluded that qualitative differences (i.e., different sources of fats) in the KD did not markedly affect seizure control (46). Nevertheless, quantitatively, a higher fat-to-carbohydrate plus protein ratio can increase seizure control clinically (2) and experimentally (25). In general, increasing the ketogenic ratio leads to improved KD efficacy (see further discussion in the section Polyunsaturated fatty acids). Numerous other possibilities are being explored in the search for the KD mechanism. Current experimental efforts are being made to determine the roles of fatty acid oxidation (47), calorie restriction (see later), neurotransmitter and neuropeptide effects (48,49), protein phosphorylation (50), and the role of glia (39,51).
Does the KD have a neuroprotective effect? Experimentally, the KD reduces the occurrence of spontaneous seizures and abnormal sprouting of dentate granule mossy fibers in the kainic acid model (53), a protective effect that was seen only if the KD was instituted within 2 weeks of the initial status epilepticus (54). Other evidence of a neuroprotective effect of the KD was found in mice with deletion of the Kv1.1 potassium channel gene; in this model, spontaneous seizures and mossy fiber sprouting also were diminished (55). The mechanism by which seizures induce neuronal damage includes mitochondrial dysfunction with the formation of injurious reactive oxygen species (ROS). The KD has been shown to increase the expression of mitochondrial uncoupling proteins and thereby reduce the neuronal damage induced by ROS (56). The KD also was neuroprotective against the damaging effects of kainic acid seizures by inhibiting caspase-3–mediated apoptosis (57).
In summary, the KD has a long and effective history of seizure control in children whose epilepsy is refractory to standard AEDs. A wealth of exciting, ongoing research is designed to unravel the mechanism of the KD and thereby improve its clinical effectiveness. Animal studies are proceeding in parallel with clinical studies to achieve this goal.
Atkins Diet
The popularity of the Atkins Diet for weight loss is evident during a visit to any supermarket or newsstand (58). Low-carbohydrate food items line grocery store shelves, and testimonials to their effectiveness fill magazines and other media. Because the Atkins diet induces a state of ketosis by providing high fat and little carbohydrate, it is theoretically possible that the Atkins diet could enable seizure control by a mechanism similar to that of the KD. Two main differences are found between the diets (see Table 2). First, the Atkins diet does not restrict calories. Second, the Atkins diet allows large amounts of protein, which is restricted on the KD.
M Comparison of ketogenic and Atkins diets
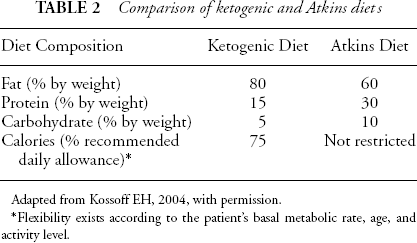
Adapted from Kossoff EH, 2004, with permission.
Flexibility exists according to the patient's basal metabolic rate, age, and activity level.
A small case series appeared recently, attesting to the effectiveness of the Atkins diet on seizure control in six patients (three children, one adolescent, and two adults), ranging in age from 7 to 52 years, with diverse types of refractory epilepsy (59). All three children and the adolescent developed ketosis, as measured by large urine ketones, whereas neither of the two adults developed large ketones. With regard to seizure control, the results were remarkable: two children and the teenager had a greater than 90% seizure reduction. Significant seizure control was not achieved in the one child lacking urinary ketosis and in either adult. None of the patients had significant side effects, hypercholesterolemia, or excessive weight loss. All three of the successfully treated patients were able to taper their standard AEDs.
Although this is a small, uncontrolled trial, it raises the possibility that the Atkins diet may be beneficial for children with medically refractory epilepsy. Children who withdraw from the KD usually do so because of poor tolerability or the family's inability to maintain the rigorous dietary regimen (11). Therefore a dietary regimen that increases palatability through less-restrictive protein and calorie requirements might enhance patient compliance. The Atkins diet seems to work better in children, as does the KD. The Atkins diet was less encouraging in adults with epilepsy, but larger-scale studies are necessary.
The observation that both diets induce ketosis and attenuate seizures may have implications in terms of understanding the mechanism, and the positive response to the Atkins diet may provide clues to the mechanism of the KD. The relative roles of ketosis and calorie restriction must be elucidated. No animal studies of the Atkins diet have appeared.
Calorie Restriction
The original idea for a KD was derived from the beneficial effect that fasting had on seizures, and this observation has been verified in the modern setting (60). This approach, obviously, is impractical except for very short-term use. Nevertheless, the health benefits of modest calorie restriction are becoming increasingly clear and include an increased life span, reduced risk of cancer and cardiovascular disease, and amelioration of the degenerative affects of aging (61,62). Restriction of calorie intake also can be neuroprotective (63,64). In rats receiving kainic acid, expected learning deficits and histologic damage are reduced on a calorie-restricted diet (65), possibly as a result of reduced oxidative stress. It is not known how calorie restriction prevents detrimental degenerative affects of aging or how a combination of environmental enrichment and calorie restriction delays their occurrence. Multiple genes that enhance neuronal survival and plasticity are likely to be involved (66). Reliable data on calorie limitation in humans with epilepsy is lacking, but it is worth considering the possibility that calorie restriction can limit seizure occurrence.
Children on the KD are typically restricted to 75% of the recommended daily allowance for their ideal weight and height. This calorie level allows linear growth but prevents significant weight gain (67). Calorie restriction may facilitate ketosis and exert an antiseizure effect, independent of ketosis. In individuals on the KD, blood glucose levels are usually in the lower end of normal, but patients are rarely hypoglycemic (2,68). Therefore a metabolic adaptation occurs in response to the KD to maintain relative euglycemia. The main clinical question, as yet unanswered, is whether calorie restriction can reduce the seizure burden independent of ketosis.
Several recent animal studies have provided evidence that calorie restriction alone can reduce seizure occurrence. The effect of calorie restriction on seizure threshold in a model of genetic epilepsy (EL mouse) was investigated by Todorova et al. (69). EL mice typically develop seizures at several weeks of age. However, by either restricting calories to about 85% of normal or providing a KD, seizure onset in EL mice was delayed by several weeks. The authors postulated that calorie restriction may underlie the mechanism of the KD, either from ketosis or hypoglycemia (69). Most likely, a combination of the two factors is occurring—the brain may metabolize ketones better under conditions of reduced glucose (70).
In another model, the threshold to seizures elicited by tailvein infusion of the convulsant pentylenetetrazole (PTZ) was measured in rats on different diets. Seizure threshold was elevated in the following order: ketogenic calorie restricted > normal diet calorie restricted > KD ad libitum > normal diet ad libitum (71). Therefore rats ingesting a normal diet restricted in calories had a higher seizure threshold than did rats on a KD without calorie restriction. Subsequently, the same group of investigators showed that even a high-carbohydrate, calorie-restricted diet (50% normal calories) afforded PTZ seizure protection, similar to that seen in a less severely calorie-restricted KD (90% normal calories) (72). In a study previously cited, calorie restriction and the KD were equally effective in suppressing dentate gyrus excitability and limiting the ictal manifestations of maximal dentate activation (29).
Taken together, these data suggest that calorie restriction is sufficient to influence seizure threshold and may augment the effects of KD treatment. Despite the beneficial effects of calorie restriction, it must be noted that several investigators reported favorable effects of the KD by using protocols that were not calorie restricted (26,57,73). Therefore the explanation for the seizure-protective benefit of calorie restriction is likely to be as complex as that for the KD itself, involving a complicated interplay between metabolic factors and excitability mechanisms.
Polyunsaturated Fatty Acids
Fatty acids play a critical role in nervous system development. The essential fatty acids, especially the long-chain polyunsaturated fatty acids (PUFAs) of the ω-3 class (as found in certain fish oils), are necessary for the development of normal retinal and neuronal membranes, as well as for subsequent normal behavior and cognition (74,75). Deficiencies of PUFAs lead to cognitive, behavioral, and structural brain abnormalities. Consumption of ω-3 PUFAs may even counteract the degenerative effects of Alzheimer disease (76). In addition to their role in brain development, fatty acids exert important modulatory effects on cellular excitability and receptor-mediated signaling pathways (77). Elegant studies have shown that PUFAs reduce excitability of cardiac muscle cells by inhibiting voltage-dependent sodium channels and L-type calcium channels, thereby stabilizing the membrane and resulting in an antiarrhythmic action (78).
PUFAs also exert important modulatory actions on neurons: they decrease or increase their firing rates, alter neurotransmitter release, and modulate synaptic responses (79). PUFAs reduce neuronal sodium and calcium currents and inhibit or activate potassium channels (80,81). When the PUFA linoleic acid (18:2 ω-6) was administered before kainic acid–induced seizures, animals were protected against status epilepticus and subsequent excitotoxic hippocampal cell death. Arachidonic acid (20:4 ω-6) inhibits voltage-gated sodium channels and shifts the sodium inactivation curve to more hyperpolarized potentials (82), resulting in suppressed neurotransmitter release and reduced repetitive neuronal firing, similar to the actions of the standard AEDs, phenytoin (PHT) and carbamazepine (CBZ). Arachidonic acid also is neuroprotective against kainic acid seizures (83). Another PUFA, eicosapentaenoic acid (20:5 ω-3), hyperpolarizes hippocampal CA1 neurons, raising their threshold for action-potential generation and reducing their baseline and convulsion-induced firing rates. It is likely that PUFA effects are regionally specific and may vary according to developmental age.
In vivo, a mixture of PUFAs of the ω-3 and ω-6 classes, in a specific ratio, raised seizure threshold in several experimental models (84). In another series of experiments using a cortical-stimulation model of seizures, long-chain PUFAs produced a transient elevation in seizure threshold, which was correlated with dynamic changes in serum PUFAs concentration (85).
Given the extensive evidence for a neuromodulatory role of fatty acids on cortical hyperexcitability, it is tempting to speculate that this class of compound might be used clinically (86). The classic KD consists largely of saturated fatty acids. In human volunteers, a KD enriched with PUFAs resulted in higher levels of circulating β-hydroxybutyrate than a standard KD (87). In a case series of nine children on the classic KD, increased serum levels of PUFAs were found, and the level of at least one PUFA (arachidonic acid) correlated with improved seizure control. Those findings are of interest because of the excitation-reducing effects of arachidonic acid previously described (88). It is notable that valproic acid (VPA), a commonly used broad-spectrum AED, is itself a short-chain fatty acid.
Because the KD provides an abundance of lipids, a reasonable hypothesis is that the hyperlipidemia induced by the diet diminishes seizure susceptibility by altering the lipid composition of neuronal or glial membranes, through an effect on membrane protein mobility, ionic channel function, or some metabolic mechanism. It is uncertain which, if any, lipid components are essential during ketosis. Discontinuation of the KD rapidly results in seizure recurrence, while lipids remain elevated, however. Perhaps lipids of certain chain lengths or degrees of saturation could exert an effect on seizure control. In rats, seizure control correlates with a higher ketogenic ratio, with maximal protection against PTZ seizures at a fat-to-carbohydrate plus protein ratio of 6:1 (25). Humans would not be able to tolerate such a high volume of fat.
Although clinical dietary trials demonstrating that PUFAs effectively reduce disorders of cardiac excitability were promising (79), few analogous attempts have been made to test PUFAs for seizures, in either patients or animal models. A single study has attempted a clinical trial of a PUFA-enriched diet in patients with epilepsy (89). A daily ω-3 PUFA supplement was given to five institutionalized patients with epilepsy. The supplement, ingested as a bread spread each morning for 6 months, consisted of 65% ω-3 PUFAs. No rationale was provided as to how this concoction was devised. Seizure frequency before the trial ranged from 1 to 14 seizures per week. After the PUFA trial, seizure counts ranged from 0 to 1 per month. Although intriguing, this study has numerous, serious limitations. The study was not blinded or controlled, involved only five patients (16 others were enrolled, but refused to eat the supplement), and no uniformity of seizure type, seizure etiology, seizure history, concurrent AED therapy, and many other variables was ensured. No attempt was made to vary the composition of the supplement, and no crossover to a regular diet occurred. Therefore these results raise the possibility that PUFA supplementation may improve seizure control; but before concluding a positive affect, these concerns must be addressed in a much larger, well-controlled prospective study. A blinded, controlled clinical of PUFA supplementation in adults with epilepsy is currently under way (E. Bromfield and B. Dworetzky, personal communication).
In conclusion, PUFAs play important roles in brain development and in the modulation of neuronal excitability. Exploration of their possible function in epilepsy treatment is just now beginning. It is possible that a “PUFA diet,” analogous to the existing KD, could become a feasible mode of therapy against epilepsy.
Dietary Factors that May Worsen Seizures
Although this review has focused on dietary manipulations that may alleviate seizures, it must be acknowledged that some diets or dietary constituents may worsen seizure control. Glutamate, the primary excitatory neurotransmitter, is clearly epileptogenic (90); excess intake of glutamate (in particular, monosodium glutamate) has been implicated in seizure exacerbation. Stimulants, such as caffeine, also have been reported to worsen seizures, which probably is due to blockade of the adenosine α1 receptors—activation of which is anticonvulsant (91). Alcohol ingestion lowers seizure threshold, in both the short and long term (92). Some dietary practices include particular foods that can cause seizures [e.g.,betel nuts (93)]. A wide variety of herbal remedies can exacerbate epilepsy, even some that are touted to suppress seizures (94). This list obviously is incomplete, and it is certainly simplistic to suggest a direct link between each of these food substances and epilepsy, as undoubtedly more complex regulatory mechanisms are involved. However, the physician must be aware of the potential for certain foods to exacerbate seizures.
Conclusions
Aside from offering common sense (i.e., “eat a healthy, well-balanced diet”), is it possible to offer epilepsy patients any specific advice regarding nutrition? Unfortunately, our knowledge about the relation between nutrition and epilepsy is in its infancy. Aside from the KD, nutritional modalities to treat epilepsy are premature. Nevertheless, as indicated in this review, several potential treatment adjuncts are on the horizon. Further basic research is necessary before clinical trials are undertaken with these prospective treatments. However, dietary alterations comprise an intriguing and novel approach to epilepsy treatment. Patients should be cautioned not to initiate any of these diets without medical supervision.