
Abstract
Commentary
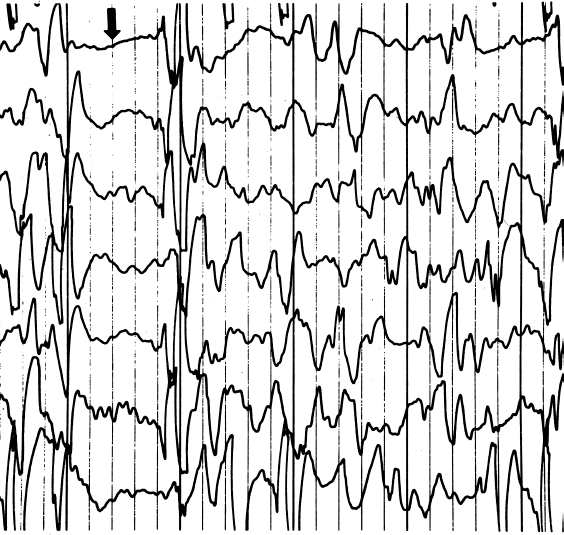
EEG of a child with West syndrome/infantile spasms. Infantile spasms are coupled to a characteristic age-dependent EEG pattern called hypsarrythmia. The pattern consists of chaotic spikes, polyspikes and slow waves, interspersed with burst-suppression periods (see arrow).
While the basis for most infantile spasms is unknown, findings in worms, mice, and humans suggest that it can be caused by genetic mutations that result from defects in neurotransmitter release (3–5) or impair neuronal migration (6). More than 60 distinct disease-associated mutations in seven X-linked mental retardation syndromes are known. One of these mutations occurs in the Aristaless-related homeobox gene (ARX) that maps within a locus on Xp22 (7,8). Seizure disorders associated with ARX mutations include West syndrome, X-linked myoclonic epilepsy, Partington syndrome, and nonsyndromic X-linked mental retardation (8,9). Infantile spasms can also result from infections, head trauma, or hypoxic–ischemic injury. The heterogeneous etiologies of infantile spasms point to links between pediatric seizures and disruptions in developmental events or neural pathways. To study the mechanisms by which the underlying lesion, brief seizures, or hypsarrhythmia disrupt developmental processes and lead to permanent cognitive deficits, improved animal models of West syndrome are needed (10). Now there are two mouse genetic models for West syndrome that show hypsarrhythmia, profound reductions in forebrain GABAergic interneurons in the adult brain, and concomitant cognitive deficits.
ARX belongs to a family of homeodomain-containing transcription factors that enhance or repress different genes in various tissues. Conserved domains, such as the aristaless domain and the prd-like class homeodomain, regulate the expression of many genes involved in embryo patterning, possibly explaining why infants with mutations in these homeodomains show catastrophic brain malformations (11). A third domain contains repeating polyA tracts (translated into alanines). Common mutations increase the polyA tracts and with them, the repression of target genes (11). Four polyA tracts are in the gene, including a mutation-prone hot spot in the second exon. The length of the polyA tracts couples with the severity of the disorder: shorter expansions of one to three alanines cause mental retardation (12), while longer expansions, which create seven extra alanine residues, cause infantile spasms and West syndrome (13). The longest mutation expands to 27 alanine residues and causes early infantile epileptic encephalopathy with a suppression burst pattern (14).
Postmortem analyses of a patient with an ARX mutation showed that abnormally few GABAergic neurons are located in the cerebral cortex, but many appear in the white matter and subventricular zones—regions that do not normally contain interneurons (6). Kato and Dobyns proposed that deficiencies in GABAergic interneurons might cause infantile spasms, possibly by deregulating a common ontological pathway for interneuron development (15). Their hypothesis, termed interneuronopathy, attempts to unify the clinical symptoms with neuropathological abnormalities. Until recently, scientists lacked the ability to directly test the interneuronopathy hypothesis because Arx null mice, which also have disturbances in fore-brain interneuron distributions, die at birth (7,16). Thus, the Arx null mice do not live long enough to examine their EEG patterns or behavior, making it impossible to verify if the deficits in interneuron positioning cause motor spasms and hypsarrhythmia.
Currently, two independent groups, reviewed here, have created nonlethal Arx deficits in mice and have demonstrated how ARX mutations cause dysfunctional interneurons. Marsh et al. created an allele of Arx flanked by lox-p sites. The usefulness of this construct is that any allele flanked by these sites (i.e., floxed alleles) is excised in the presence of the enzyme Cre-recombinase. The floxed mice were mated to a mouse that expresses Cre-recombinase under the control of the Dlx5/6 enhancer element which is active in calbindin interneuron subtypes. Neither transgene alone causes a defect, however, when the two strains are mated, the male offspring become Arx deficient (Arx-/Y;Dlx5/6 CIG ) and the female offspring have only one functional copy of Arx (Arx-/+;Dlx5/6 CIG ).
EEG abnormalities were found in the Arx-deficient mice on postnatal days 14–17. All of the Arx-/Y;Dlx5/6 CIG males, and a majority of the Arx-deficient female mice showed spontaneous seizures. EEG abnormalities were further evaluated with continuous video and intracranial EEG recordings for up to 1 month when the mice were 3- to 4-months old. The male Arx-deficient mice also showed reduced theta and delta activity. Deficits in various GABAergic interneuron populations in these mice correlate with the EEG findings.
A second study by Price et al. examined the effects of Arx mutations consisting of the triplet repeat expansion, which is found in multiple human patients (13). They created a trans-gene with repeating stretches encoding for the amino acid, alanine, in critical exons of the Arx gene. In exon 2 of the human ARX gene, the polyA tract is normally 10 codons long, but an insertion of seven repeats of the trinucleotide sequence, guanine–cytosine–guanine (this DNA trinucleotide is the codon for the amino acid alanine), occurs in West syndrome. The result is a longer tract of 23 alanines in the protein. This mutation is designated ARX(GCG)10+7 . Price et al. created a similar insertion mutation by targeting a construct of the mouse Arx gene that causes expansion of the initial polyA tract in exon 2. They designated this mouse line Arx(GCG)10+7.
Postnatal Arx(GCG)10+7 mice showed infantile motor spasms, spontaneous seizures, and EEG abnormalities, including sharp-spike, slow-wave transients; electrodecremental EEG events; and increased high-frequency background rhythmic activity. They also demonstrated cognitive impairments. In addition, the mutants displayed reduced anxiety-like behaviors on a light/dark exploration test, compared to wild-type mice. When tested in a pavlovian associative learning paradigm that pairs a tone with a brief foot shock, the mutant mice show a deficit in learning and memory. A final test described a measure of social interactions with other mice. When confronted with an unfamiliar mouse in their home cage, most mice actively investigate the interloper. However, the Arx mutant mice retreat, an anomalous social behavior.
Immunohistochemical studies evaluating the distribution of ARX-expressing GABAergic interneurons showed that they were almost entirely missing in the cerebral cortex, the hilus of the dentate gyrus, and the striatum of the mutant mice. Surprisingly, interneurons show normal distributions in the parietal cortex, whereas the overall decrement in GABAergic cortical interneurons in the Arx(GCG)10+7 mice is approximately 50%. Moreover, the mice have fewer calbindin+ and neuropeptide Y+ interneurons, but normal distributions of parvalbumin+ and calretinin+ interneurons. Though it seems likely that reduced inhibitory neuron numbers in the cerebral cortex and hippocampus account for the clinical EEG features, hypsarrhythmia also could be related to a region and area-specific cortical circuit anomaly. With these new mouse models, it may now be possible to map the functional neural circuits underlying age-related EEG anomalies.
Of what consequence are the trinucleotide expansions for the transcriptional activity of ARX protein in the immature brain? ARX normally localizes to the nucleus, but it becomes cytoplasmic in the mutant mice (17). Expansion mutations in other transcription factor genes can cause proteins to misfold, resist degradation, and form aggregates in the cytoplasm. Such changes could cause subtle impairments in transcriptional regulation and neuronal development that may have profound clinical implications, as the expression of over 84 downstream target genes is altered by disrupting Arx (18). Now the search is on for interneuron-specific genes responsible for the development of interneuronopathy and for how such gene expression is changed in ARX mutations (19). The pioneering work presented in these two papers allows researchers to embark on an extremely promising adventure across uncharted territory in the molecular biology of West syndrome.