
Abstract
One by one, mutation-containing mendelian genes that cause monogenic juvenile myoclonic epilepsies (JME) and single nucleotide polymorphisms (SNP)-susceptibility alleles that increase risks for nonmendelian complex JME should fall to the power of molecular genetics. Of 15 chromosome loci, 3 mendelian genes (α1-subunit of the GABAA receptor [GABRA1], chloride channel 2 gene [CLCN2], and Myoclonin1/EFHC1) and 2 SNP-susceptibility alleles of putative JME genes in epistases (bromodomain-containing protein 2 [BRD2] and connexin [Cx]-36) have been identified, so far. Antiepileptic drugs now can be designed against the specific molecular defects of JME.
Juvenile myoclonic epilepsies (JME) are primarily genetic in origin. One by one, mendelian epilepsy genes and their mutations that cause monogenic JME will unravel to linkage analyses and positional cloning, using short tandem repeat polymorphisms (microsatellites). At the same time, complex, nonmendelian JME with its susceptibility alleles should be sieved by association analyses and transmission linkage disequilibrium, using single nucleotide polymorphisms (SNPs).
In dominantly inherited epilepsies, the ideal situation for research purposes is to have one large family or a set of large families (each with a minimum of nine affected members over at least three generations) in order to independently prove linkage to the same chromosome site (logarithm of odds [LOD] score >3.3 at theta=0 during genome-wide scan, using microsatellites spaced 1cM apart, or LOD score >3.6 at theta=0 during genome wide scan using microsatellites spaced 0.1 cM apart—see Table 1). In recessive epilepsy traits, six affected offspring belonging to consanguineous matings in one large kindred usually will reveal homozygosities that point to the chromosome site supported by significant Lod scores. Such approaches to mendelian epilepsies have successfully identified mutation-containing genes for autosomal dominant JME (1–3), autosomal recessive Lafora disease (4), and Unverricht Lundborg disease (5).
Identifying Chromosome Loci and Epilepsy Genes
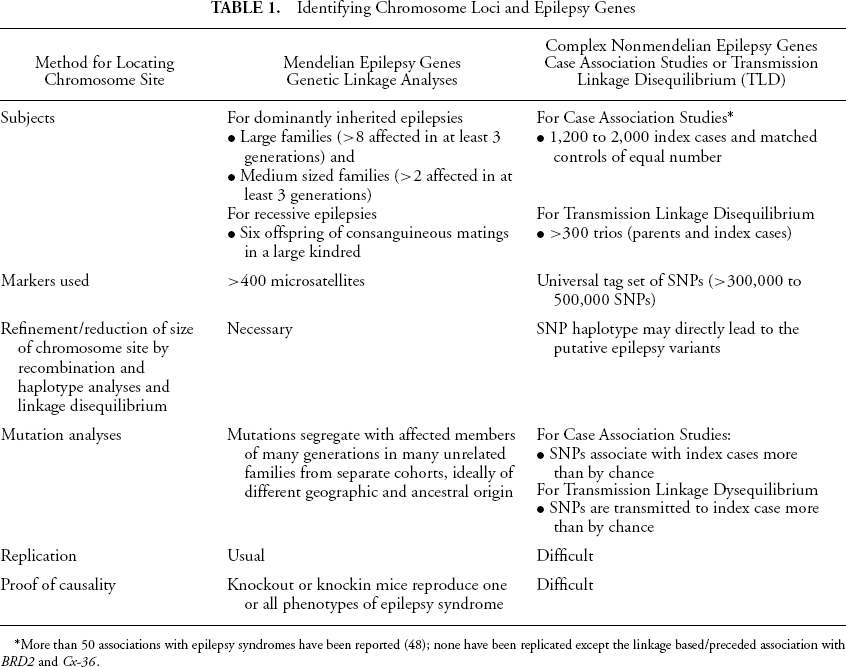
More than 50 associations with epilepsy syndromes have been reported (48); none have been replicated except the linkage based/preceded association with BRD2 and Cx-36.
In epilepsies that are clearly heritable yet do not follow any of the known modes of inheritance, it is thought that multiple genes interact (epistases) and produce epilepsy phenotypes. Because the heritability effects of such genes are significant enough to contribute to the epilepsy phenotype but only produce a minor proportion of the whole trait, they are undetected by traditional linkage analyses. To detect these multiple-gene epilepsy phenotypes, a large number of patients and controls (i.e., 1,200 to 2,000 unrelated index cases and an equal number of age, sex, and racial/geographically matched controls) would need to be enrolled in a genome-wide scan that uses tagging sets of SNPs (300,000 to 500,000 SNPs) and is aided by the 2005 International Haplotype Map (HapMap). This protocol statistically associates an SNP variant with a specific type of epilepsy. It allows construction of tag SNP haplotypes, which often travel together through generations of recombinations and could directly identify the many common DNA variants that underlie susceptibility to epilepsy (6). Such linkage disequilibrium-based case association studies or transmission linkage disequilibrium analyses of trios have not been executed in genetically complex epilepsies. Instead, SNP susceptibility alleles in previously linked chromosome sites have been associated with small cohorts of epilepsies, as will be discussed here.
Table 2 lists 15 JME chromosome loci that have been genetically linked to JME by genome-wide linkage analysis or by linkage analyses that tests specific candidate regions (1). Two mutation-containing ion channel genes, α1-subunit of the GABAA receptor (GABRA1) (2), and chloride channel 2 gene (CLCN2) (3), as well as one non-ion channel gene, myoclonin1/EFHC1(4) have been identified as putative JME genes. Mutations in the two ion channel genes have, so far, been unique to a handful of families (1,2). In contrast, mutations in myoclonin1/EFHC1 are found in 9% of consecutive JME cases from neurology clinics, including singletons, sporadics, and surprisingly also in juvenile absence epilepsy and temporal lobe epilepsy (7,8). In the last 4 years, much progress in JME research has concerned the discovery of the three mendelian JME genes mentioned and the genetic association of SNPs of bromodomain-containing protein 2 (BRD2) (9,10) and of Connexin (Cx)-36 (11) with genetically complex JME. These are the subjects of this review.
Chromosome Loci and Genes Identified in JME
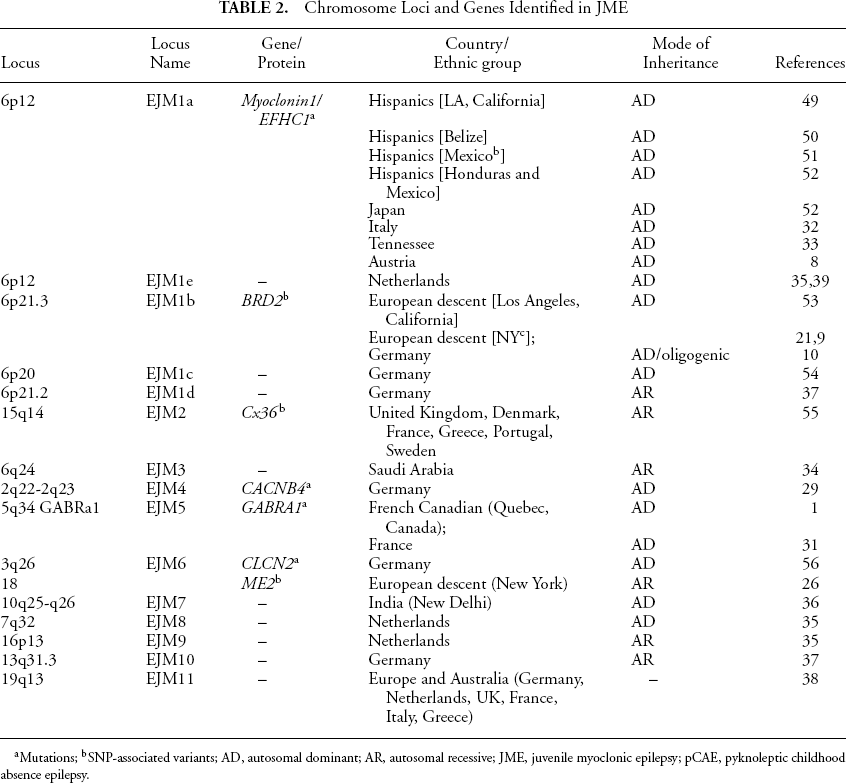
Mutations
SNP-associated variants; AD, autosomal dominant; AR, autosomal recessive; JME, juvenile myoclonic epilepsy; pCAE, pyknoleptic childhood absence epilepsy.
Phenotypes of JME
JME is the most common form of idiopathic generalized epilepsies and the most common cause of primary grand mal seizures. JME accounts for 12% to 30% of all epilepsies in hospitals and clinics (1215), while a house-to-house population survey estimates and undercounts JME at 3% (16). Two major and two minor subsyndromes form the majority of JME (17). The first major subsyndrome is classic JME with no absences and accounts for 72% of all JME patients. Awakening myoclonias start at 13 to 15 years, and grand mal convulsions appear 2 years later. Absence seizures are not present in probands and do not appear as the sole phenotype in affected nonproband family members. The second major subsyndrome, childhood absence that evolves to JME, affects 18% of JME cases. Pyknoleptic absences with 3 Hz spike and wave complexes and 4 to 6 Hz spike or polyspike waves complexes persist into adolescence, when myoclonias and grand mal convulsions start. Nonproband affected family members can have absence only, grand mal only, or absence and grand mal but rarely have JME. The two minor JME subsyndromes consist of adolescent myoclonias and grand mal convulsions with absences that start during adolescence or after 18 years of age (7%). Even more rare (3%) are astatic drop seizures, which appear during adolescence, together with myoclonic and grand mal seizures (17).
Neuropathology of some patients with JME reveals increased number of and diffusely distributed dystopic neurons in gray matter stratum moleculare and subcortical white matter of brains (18). Quantitative high-resolution voxel-based MRI detects such JME neuropathology as significantly larger, thicker, and increased cerebral cortical gray matter in mesial frontal lobes (19,20).
Susceptibility SNPs for Complex Nonmendelian JME
After refining and narrowing a candidate chromosome locus for JME, the intent is to identify mutations that are absent in control populations and that segregate with affected members of multiplex and multigenerational families. In two candidate loci for JME, no mutations were found to segregate with affected family members. Instead, SNPs contained within putative JME genes were statistically associated with a small number of probands. These probands belong to small- or medium-sized multiplex or multigenerational JME families that had originally been genetically linked to chromosome 6p21.3 (human leukocyte antigen [HLA] region) and associated with HLA alleles, as in New York families of European origin (21,22), and to chromosome 15q14, as in JME families from Sweden and United Kingdom (23).
In 1996, Greenberg and colleagues reported that JME families from New York had a locus within the HLA region, between HLA-DP and HLA-B loci (21,22), expressing both grand mal seizures on awakening and with JME (24). Both Greenberg et al. and Obeid et al. from Saudi Arabia noted that DR13 was significantly associated with JME families (25). These two HLA-association studies suggest that a second JME gene (i.e., aside from the 6p12 gene) may be present within the HLA complex of chromosome 6p. JME families from Los Angeles, Belize, Mexico, Honduras, and Germany do not have significant association with HLA alleles and eventually were shown to have separate loci in chromosome 6p21.2, 6p20, and 6p12.
In 2003, Pal et al. showed that SNPs within the BRD2 (RING3) gene in chromosome 6p21.3 might be susceptibility alleles (odds ratio 6.5) for autosomal recessive JME families from New York (9). Linkage disequilibrium found two strongly JME-associated SNP variants in the promoter region of BRD2 and a common variant haplotype in over 50% of 20 probands from families that had produced positive LOD scores for 6p21 during linkage analyses (9).
Greenberg and colleagues (26) again used both case-control and family-based association studies in identifying malic enzyme 2 (ME2)-centered, nine-SNP haplotype in chromosome 18q. When present in the homozygous state, the ME2-centered nine-SNP haplotype increased the risk for idiopathic generalized epilepsy (odds ratio 6.1). The results of Pal et al. and Greenberg et al. suggest, but do not prove, that the BRD2 gene in 6p21.3 epistatically interacts with the ME2 gene in 18q to produce JME (9,26). In 2006, Lorenz et al. (10), tested whether alleles of BRD2 also conferred susceptibility to the EEG photoparoxysmal response in JME/idiopathic generalized epilepsy families from Germany. Allelic and haplotypes associations were found between the photoparoxysmal response and six BRD2 polymorphisms; however, the polymorphisms appeared to be protective against, instead of increasing risk for photoparoxysmal response.
Like the New York group, Mas et al. (11) found susceptibility SNPs in JME families from the United Kingdom and Sweden. They sequenced the gene Cx-36 in 29 JME probands previously linked to the 15q14 locus and did not find any epilepsy-causing mutations that segregated with affected members. Instead, a case-control study found significant association between JME and the c.588T polymorphisms within exon 2 of Cx-36. With significant differences in both alleles (p = 0.03) and genotype (p = 0.017) frequencies, subjects with the T/T genotype at position 588 had a significantly increased risk for JME (odds ratio 4.3) compared to those with a C/C genotype. HAP2, a haplotype containing c.588T, was significantly associated with JME (p = 0.03) (8). Hempelmann and colleagues replicated association of c.588T (dbSNP:rs3743123, S196S) within Cx-36 of 247 German JME patients versus 621 controls (27). They observed an increase in homozygotes (13.4%) in JME compared with controls (8.7%), suggesting the allele increased the risk for JME in the homozygous state or in an autosomal recessive form of transmission.
These susceptibility polymorphisms (i.e., BRD2 in chromosome 6p21.3; Cx-36 in chromosome 15q14; and ME2 in chromosome 18) are hypothesized to act in epistases as minor epilepsy genes in the homozygous state, necessary but not sufficient, alone, to produce a JME phenotype. The challenge with the SNPs is to show what changes in cell function are altered so as to produce the JME phenotypes. None so far have been tested. A second, more formidable challenge is proving causality (see Table 1). It is a difficult challenge to insert SNPs of putative JME genes in a bacterial artificial chromosome (BAC) vector—together with other SNPs of putative JME genes that they are hypothesized to interact with, in epistases—and then transgenically reproduce one or all of the JME phenotypes. The BAC gene targeted approach rarely has been used to prove that interacting polymorphisms can cause a given phenotype, as Wu et al. showed for the “trans” polycystin gene in the mouse model of polycystic kidney disease (28).
Genes for Monogenic JME
As mentioned, three genes—GABRA1 in 5q34; CLCN2 in 3q26; and myoclonin1/EFHC1 in 6p12—have mutations that segregate with affected members of multigenerational families and have possible mechanisms for producing JME phenotypes, which satisfy two of three criteria for a gene to be called a putative JME-causing gene. The third criterion is the generation of a knockout or knockin mouse model that replicates one or all of the electroclinical and neuropathological phenotypes of JME. Amongst the three putative JME genes, only myoclonin1/EFHC1 so far has met the third criterion (Suzuki T, Delgado-Escueta AV, Yamakawa K, unpublished data, 2006). Major mendelian JME genes are considered necessary and alone sufficient to produce a JME phenotype.
Calcium channel β4 subunit (CACNB4) is not considered a putative JME gene because its mutation did not segregate in affected family members, was found in only one member of a JME family from Germany, and has not been replicated (29). Chloride channel 2 gene (CLNC2) mutations segregated in affected members of one large German JME family; mutations also segregated with affected members of two other families with idiopathic generalized epilepsies (2). Mutations in CLCN2 also have been observed in rare families from Brussels, Belgium (30). Thus far, GABRA1 receptor gene has been confirmed in one absence family from France and, recently, in a large French Canadian JME family (1,31).
Myoclonin1/EFHC1 missense, nonsense, frameshift-deletion, and deletion mutations now have been found in 6 separate cohorts, including 20% of the first cohort of 30 Hispanic families from California and Mexico and 9% of a second cohort of 44 clinic index cases from Mexico and Honduras (7). In this second cohort, mutations were found in 3 families, 2 singletons, and a true sporadic with a de novo mutation. In the third cohort of 72 JME families from Japan, only 3% carried a missense mutation in transcript A and a deletion mutation in the 5'UTR promoter region of myoclonin1/EFHC1 (7). The fourth cohort consisted of 25 JME families from Italy; 20% showed missense mutations in Myoclonin1/EFHC1 (32). The fifth cohort consisted of 54 JME families from Tennessee; only one mutation, R221H, was found (33). In the sixth cohort myoclonin1/EFHC1 was used as a probe to define how broad its phenotype was in 61 patients from Austria with various idiopathic epilepsies and in 372 patients with temporal lobe epilepsy (8). Three novel heterozygous missense mutations and one possibly pathogenic variant in the 3'UTR (2014t > c) were found in two patients with juvenile absence, an unclassified epilepsy syndrome, and in one patient with JME. The I174V missense mutation also was found in one patient with temporal lobe epilepsy. Thus, mutations in myoclonin1/EFHC1 are relatively more common in Hispanic JME populations from California, Mexico, and Honduras and in Caucasians from Italy and Austria. Such mutations are rarely to infrequently seen in Japan and Tennessee. Mutations in myoclonin1/EFHC1 also may underlie idiopathic juvenile absence and temporal lobe epilepsies. JME-causing mutations or susceptibility SNPs have not yet been discovered in the eight other reported JME chromosome loci (i.e., 6q24 in Saudi Arabia families (34); 16p13 and 7q32 in Dutch photosensitive myoclonic families (35); 10q25-26 grand mal locus in New Delhi families (36); 13p in photosensitive German families; 6p20 and 6p21.2 in German families (37), and 19q13 in European and Australian families (38) or the 6p12 locus of Dutch families (39).
Myoclonin1/EFHC1 in mouse hippocampal primary culture neurons induces apoptosis that is significantly lowered by mutations and suppressed by SNX-482, an antagonist of R-type voltage-dependent calcium channel Cav2.3. In patch clamp analysis, Myoclonin1/EFHC1 specifically increases R-type calcium currents, which are reversed by JME mutations (3). In addition, myoclonin1/EFHC1 is involved in cell division. During mitosis, both C- and N-terminus EGFP tagged myoclonin1/EFHC1 associates with mitotic structures. In interphase cells, myoclonin1/EFHC1 localizes with the centrosome, interpolar microtubules, and spindle poles (40).
Is JME a Ciliopathy?
Myoclonin1/EFHC1 may also be involved in cell migration. Ikeda et al. demonstrated that EFHC1 is the mammalian orthologue of Chlamydomonas polypeptide and the Rib72 gene, which is found within protofilament ribbons of motile cilia (41,42). EFHC1 expression is most robust in murine ventricular regions and cerebral aqueduct where cilia is present (43,44). Because CSF flow is driven by motile cilia of ependymal cells and establishes a chemical gradient within subventricular zones that is required for migration of neuroblasts (45), King predicts that EFHC1 mutations will produce dominant negative effects on ciliary functions, impair the chemical gradient in the subventricular zone, and disturb migration of neuroblasts (43). This hypothesis could provide another explanation for the thick cerebral cortex seen on MRI and for the dystopic or displaced neurons present in stratum moleculare and white matter seen on neuropathology of some JME patients.
Blast searches of Drosophila databases, using the human protein sequences, identified two fly homologs of myoclonin/EFHC1—CG11048 for myoclonin1/EFHC1 in 6p12 and CG8959 for myoclonin2/EFHC2 in Xp11.4 (46). In 2005, Gu et al. (47) also cloned the brain-expressed paralog myoclonin2/EFHC2 located in chromosome Xp11.3. These authors reported a tentative association between S430y in exon 9 of myoclonin2/EFHC2 in 97 male JME patients from Germany, providing a possible explanation for increased maternal inheritance in JME.
Conclusion
JME accounts for at least 3% to as much as 30% of all epilepsies and is primarily genetic. Depending on racial/ethnic and geographic origin, JME trait is transmitted as a mendelian dominant or recessive trait or as complex oligogenic traits. As reviewed, 15 chromosome loci, 3 mutation-carrying major JME genes, and 2 SNP susceptibility genes in epistases have been identified so far. Among mendelian JME genes, Myoclonin1/EFHC1 is the most common and is involved in cell division, apoptosis, and survival as well as postsynaptic calcium homeostasis via R-type voltage-dependent calcium channel function. There is great potential for improving human health if antiepileptic drugs are designed to counter major genes that cause JME. Drugs designed against genetic epilepsies generally have had broad antiepileptic actions, working against both idiopathic and lesional epilepsies.