
Abstract
The mechanisms that transform a normal brain to an epileptic one are not fully understood. Interleukin-1 beta (IL-1β) contributes to neuronal degeneration observed in several neurological disorders and recently has been implicated in neuronal injury that may accompany the process of epileptogenesis. This review presents the hypothesis that IL-1β may contribute to the development of epilepsy via several mechanisms, including classical effects on neuronal survival and transcription pathways; novel rapid effects on receptor-gated ion channels; and long-lasting effects on expression of selective gene families. Thus, evidence that IL-1β actions in epilepsy can be independent from the neurotoxic effects of this cytokine is presented.
An understanding of the role of inflammatory processes, particularly of the cytokines, in human neurological diseases has been evolving. For example, inflammatory processes are now considered key contributors to acute and chronic neurodegenerative disorders, such as ischemic stroke and Alzheimer's disease (1). Insight into the many faceted roles of the cytokines, especially of interleukin-1 beta (IL-1β), in the initiation and progression of neuronal dysfunction has been gained from studies of the evolution of epilepsy. In respect to the pathophysiology of the epilepsies, clinical studies, in vivo models, in vitro molecular and imaging data, gene arrays, and the use of genetically engineered mice (2) are leading to a radically modified view of the role of cytokines in epileptogenesis, which may be germane to many other neurological diseases. Key questions include whether cytokines synthesized and released as a result of cell injury or during and after seizures contribute to the disease process itself, and if so, then how. In this short review, the evidence from studies on the mature and developing rodent brain of a role for IL-1β in epileptogenesis is examined.
Potential Roles of IL-1β in Epileptogenesis
Epileptogenesis, a transformation of a nonepileptic neuronal circuit to a seizure-generating one, is a complex process involving several key components:
Hyperexcitability of Neurons and of Neuronal Circuits
This phenomenon is particularly well defined in temporal lobe epilepsy (3). The formation of a hyperexcitable circuit may depend on the severity and duration of the initial inciting event in the case of acquired epilepsy or on the nature and extent of structural (e.g., cerebral malformation) or functional (e.g., mutated ion channel) abnormality in the case of epilepsies involving genetic causes. Cytokines released during the inciting insult may directly contribute to hyperexcitability. In addition, recurrent seizures during the epileptic phase may further release cytokines, promoting progression of the hyperexcitable state.
Seizure-Evoked Excitotoxicity
In the mature brain, the inciting event (often status epilepticus) leads to substantial loss of vulnerable neurons within the hippocampal formation; this loss is considered by many to be required for epileptogenesis (3). Cytokines and other inflammatory mediators have been shown to contribute to neuronal death (1,2). Therefore, they may participate in the epileptogenic process via augmentation or mediation of seizure-evoked cell death.
Other Putative Pro-Epileptogenic Processes
Epileptogenesis that is independent from neuronal death has been found in several genetic epilepsy models (e.g., genetic absence epilepsy rats from Strasbourg [GAERS], WAG/Rij) (4). Similarly, only minimal cell death is found after kindling (5,6) or in epilepsy evoked by experimental febrile seizures in immature brain (7,8). A role for cytokines in this epileptogenic process would not include excitotoxicity and might derive from other established or nonconventional effects of interleukin signaling.
The following paragraphs briefly review information about IL-1β contribution to excitability and excitotoxicity as well as introduce potential novel roles of this cytokine in epileptogenesis.
Studies in Adult Animal Models of Temporal Lobe Epilepsy
Temporal lobe epilepsy can be modeled in adult rodents by provoking seizures or status-epilepticus. In these models, there is evidence of neuronal death, the reactive formation of new synapses, and the molecular and functional reorganization of a previously normal circuit into an epileptic one (3). Furthermore, these models have been useful in probing the potential roles of cytokines in the epileptogenic process. Seizures and status epilepticus induced by chemical or electrical means stimulates a massive inflammatory response in the brain that consists of increased levels of cytokines, including IL-1β. Unlike the inflammatory response caused by endotoxemia—which is confined to the choroid plexus, vessels, and microglia—the inflammatory response after seizures also involves neurons and astrocytes and occurs specifically in brain regions of seizure onset and propagation. The CNS inflammatory response following seizures is rapid (within an hour) and long-lasting (2). In addition, a unique facet of the inflammatory response to seizures is the time course of induction of activators and repressors of the IL-1β receptor: when released, IL-1β binds with high affinity to the type 1 IL-1 receptor (IL-1R1) that is expressed by hippocampal neurons (9–11). In typical peripheral inflammatory reactions, the production of IL-1β is accompanied by concomitant synthesis of 100- to 1000-fold excess of the endogenous IL-1R antagonist (IL-1RA), which rapidly occludes the activation of this receptor (12). In contrast, when seizures evoke rapid production of IL-1β, IL-1RA is upregulated to a far lower extent and with a several hour delay (13,14). This finding suggests that the brain is much less effective than the periphery in inducing a crucial mechanism for rapidly terminating the effects of a sustained rise in endogenous IL-1β.
Nonconventional, Rapid Actions of IL-1β on Neuronal Excitability
Findings from in vivo experiments support a role for IL-1β signaling in hyperexcitability because IL-1β exacerbates seizures in rodents when applied intracerebrally shortly before kainic acid or bicuculline administration (15,16). In contrast, IL-1RA acts as an anticonvulsant, and in IL-1RA–overexpressing mice, seizure onset is delayed and seizure spread is strongly attenuated (16,17). In addition, seizures are reduced by blocking the formation of the biologically active form of IL-1β by selective inhibition of interleukin-1 converting enzyme (ICE or caspase-1) or by enzyme gene deletion (18).
How does lL-1β that is released during the inciting insult contribute to hyperexcitability, and how might it promote progression of the hyperexcitable state? The in vivo effects in experimental models occur within a few minutes of the application of the cytokines; therefore, they cannot be explained by the classic signaling cascade involving activation of genomic transcriptional events (12). Rather, these effects are consistent with direct actions of IL-1β on ion channels and neurotransmitter receptors.
Recent elegant studies involving the effects of IL-1β on the activity of warm-sensitive neurons in the preoptic area and the anterior hypothalamus have shown that IL-1β signaling occurs through a rapid nontranscription-dependent enzymatic pathway. This pathway includes the IL-1R1–mediated activation of the neutral sphingomyelinase (19) and the subsequent production of ceramide, which in turn activates the tyrosine kinase protein, Src. Interestingly, IL-1β enhances calcium influx into hippocampal pyramidal cells exposed to N-methyl-D-aspartate (NMDA) by phosphorylation (via Src kinase) of the NR2B subunit of the NMDA receptor complex, which is responsible for regulating its calcium permeability properties (20). Results from this study suggest that ceramide may be the second messenger of the rapid IL-1β actions on neuronal excitability. In the hippocampus, this mechanism may constitute the basis of the proconvulsant actions of the cytokines observed in the in vivo experimental models that are dependent on NMDA receptor activation (15). In addition, IL-1β inhibits glutamate reuptake by astrocytes (21) and enhances its astrocytic release via tumor necrosis factor-alpha (TNF-α) induction (22). TNF-α also can increase AMPA-receptor density at the neuronal membrane in a molecular conformation lacking the GLUR2 subunit, which results in enhanced calcium influx (23). Astrocytic glutamate release recently has been implicated in the generation of epileptiform activity (24). It is intriguing to speculate that IL-1β may play a role.
Seizure-Evoked Excitotoxicity
Specific cytokines, including IL-1β, have been shown to contribute to neuronal injury in traumatic, ischemic, and excitotoxic conditions (1), perhaps in part via enhanced excitability. Although degenerating neurons may promote and perpetuate the production of cytokines, so that a vicious cycle of neuronal injury and inflammation is activated, it is important to note that IL-1β induction following seizures always precedes the occurrence of irreversible cell death. In addition, IL-1β affects blood–brain barrier permeability and induces immune cell infiltration into the brain (1,2). These phenomena have been found to promote hyperexcitability (24,25) as well as excitotoxicity (26).
In summary, studies of epileptogenesis in adult rodent models provide compelling evidence of a role for IL-1β. However, because the cytokine promotes both hyperexcitability and excitotoxicity and because both processes take place during epileptogenesis, it is difficult to sort out what elements of the actions of IL-1β are crucial, that is, required and sufficient for its contribution to the generation of spontaneous recurrent seizures. Increased production of inflammatory molecules in the brain have been reported after fully kindled seizures (27) and in genetic models of audiogenic seizures (28)—situations in which epileptogenesis is independent from neuronal death. A role for IL-1β in the epileptogenic process in these scenarios would not include excitotoxicity and might derive from other established effects of interleukin signaling. In the next section, such roles for IL-1β in the epileptogenic process are examined on the basis of the studies involving immature animal models of seizures and epileptogenesis.
Studies in Immature Animal Models of Epilepsy: Mismatch between Excitotoxicity and Epileptogenesis
Over the past 20 years, the majority of studies that have used animal models of developmental epilepsies generally have supported the view that the crucial contribution of cytokines to epileptogenesis involves their effects on excitotoxicity. Most of these studies demonstrated that status epilepticus—provoked during the age of hippocampal development equivalent to that of the human infant (i.e., prior to postnatal day 14–15 in the rat [29])—failed to trigger the onset of epilepsy later in life. Absence of the epileptogenic process was associated with commensurate absence of significant cell death in the hippocampus (30–32) and with a lack of up-regulation of endogenous IL-1β (32), which is consistent with the hypothesis that IL-1β–mediated excitotoxicity is a key mechanism in the epileptogenetic process.
In contrast, an infant rodent model of human childhood prolonged febrile seizures recently has been defined; in this model (as is likely in humans), IL-1β intrinsically contributes to the generation of the seizures (33) and the inciting seizures do evoke epileptogenesis (8). Remarkably, excitotoxicity (i.e., cell death) does not accompany the epileptogenic process, indicating that cell death is not required for epilepsy generation (7,8,30,31). Thus, the developmental febrile seizures model dissociates the mechanisms of the pro-epileptogenic actions of IL-1β from excitotoxicity.
Fever, Febrile Seizures, and IL-1β
Fever is a systemic host response to infection, inflammation, or stress (12). Fever provokes convulsions (febrile seizures) in 3–5% of young children in the western world and in up to 14% in Japan (34). The mechanisms by which fever evokes febrile seizures are not fully elucidated, but a role for IL-1β is supported by several lines of evidence. First, IL-1β produced from peripheral and potential brain sources is involved in the pyrogenic process leading to fever (35). Indeed, levels of the cytokine have been reported to increase in CSF of children with febrile seizures (36), although these data are controversial (3739), and some studies have suggested that genetic variance that enhances IL-1β expression promotes the occurrence of febrile seizures, a notion that also has been challenged (40).
Direct evidence for the role of endogenous IL-1β in the induction of febrile seizures is provided by studies from animal models. For example, in a rodent model of febrile seizures, the ablation of the gene for IL-1R1 interferes with the ability of hyperthermia to evoke experimental febrile seizures (33). Moreover, the intracerebral application of IL-1β in immature rodents decreases the threshold for induction of experimental febrile seizures (33). It is notable that cells in the hippocampus, a region contributing to the origin and spread of febrile seizures, synthesize IL-1β in response to fever or hyperthermia (41–43). Remarkably, IL-1β is not synthesized or released during other types of experimental seizures in the immature rodent (e.g., those induced by KA). Experimental febrile seizures provoke long-lasting alterations in the expression of several intrinsic neuronal genes and promote epileptogenesis. Therefore, is it postulated that hyperthermia-evoked release of endogenous IL-1β acutely enhances neuronal excitability and leads to more enduring alterations in the transcription of specific genes, via activation of the mechanisms depicted in Figure 1.
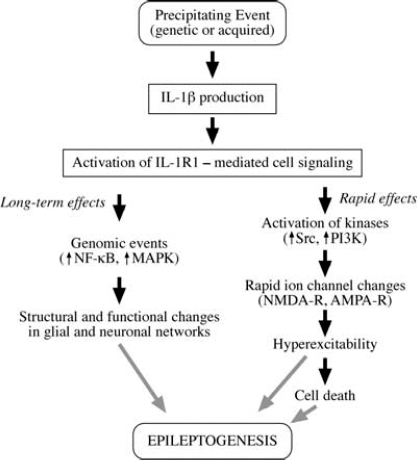
IL-1β and Epileptogenesis. This schema depicts the cascade of events that may underlie CNS actions of IL-1β after its production and release following a precipitating event. Brain IL-1β is mainly produced by glia (microglia and astrocytes); endothelial cells of the blood–brain barrier, neurons, and lymphocytes represent additional sources. Since the same cells that produce IL-1β also express IL-1 type 1 receptors (IL-1R1), it appears that this cytokine is endowed with both autocrine and paracrine actions. Activation of IL-1R1 by IL-1β can trigger rapid effects on neuronal excitability, leading to rapid ion channel changes mediated by activation of kinases, such as Src and phosphoinositol 3 kinase (PI3K) (19,20,23). These actions are likely to be responsible for the proconvulsive actions of IL-1β and may contribute to seizure-associated neuronal death. Long-term effects also may be triggered by IL-1β via transcriptional activation of NF-κB- and MAPK-dependent genes involved in structural and functional changes in glial and neuronal networks. These actions, individually or in concert, may contribute to epileptogenesis. However, available experimental evidence indicates that induction of neuronal cell death does not appear to be a prerequisite for the pro-epileptogenic actions of IL-1β.
Febrile Seizures and Epileptogenesis: The Clinical Conundrum
Prolonged human febrile seizures are associated statistically with the development of temporal lobe epilepsy (44). Retrospective (but not prospective) analyses suggest that approximately 30–70% of individuals with temporal lobe epilepsy have a history of these seizures early in life (4447). This fact makes understanding the mechanisms of epileptogenesis generated by febrile seizures an important clinical problem. As mentioned, direct evidence for febrile-seizures–evoked epileptogenesis has been provided by an animal model (8) that permits mechanistic analysis of the role of IL-1β in this process. Because epileptogenesis after experimental febrile seizures does not require excitotoxicity, other mechanisms are likely to contribute to this pathogenic process.
Figure 1 summarizes the putative pro-epileptogenic actions of IL-1β–mediated by IL-1R1 activation. These events occur during the process of epileptogenesis, regardless of whether excitotoxicity and cell death take place (as in adult poststatus epilepticus epileptogenesis) or not (as in immature brain after febrile seizures or in genetic epilepsy models).
Classical signaling cascades include the mitogenactivated protein kinases (MAPKs) and NF-κB–dependent pathways that lead to increased gene transcription. The genomic effects of IL-1 β contribute to enduring alterations in gene expression programs that may underlie the epileptogenic process and may occur even without cell death or overt structural changes. It has been postulated that the long duration of the seizure-evoked release of IL-1β (48) may account for the enduring effects of the cytokine on gene expression of receptors and ion channels (49,50).
The novel, direct actions of IL-1β on neuronal transmission strongly suggest that it may contribute significantly to lowering the threshold for neuronal excitability, thus playing a role in the onset and recurrence of spontaneous epileptic events.
Both genomic and rapid effects of IL-1β may impact cell survival as well as the consequent structural and functional reorganization of neuronal networks. However, available experimental evidence indicates that induction of neuronal death does not appear to be a prerequisite for the pro-epileptogenic actions of IL-1β.
In conclusion, the spectrum of mechanisms by which IL-1β may contribute to epilepsy is increasing as refinement of tools permits dissection of not only overt effects (e.g., cell death) but also of subtle and pervasive alterations of cellular function. Pharmacological investigations using the available tools to block endogenous IL-1β production and/or its cell signaling after the inciting event in experimental models of acquired epilepsies are required to define better the role and relevance of this cytokine in epileptogenesis (15,16,18).