
Abstract
Vitko I, Chen Y, Arias JM, Shen Y, Wu XR, Perez-Reyes E
J Neurosci 2005;25(19):4844-4855
Sequencing of the T-type Ca2+ channel gene CACNA1H revealed 12 nonsynonymous single nucleotide polymorphisms (SNPs) that were found only in childhood absence epilepsy (CAE) patients. One SNP, G773D, was found in two patients. The present study reports the finding of a third patient with this SNP, as well as analysis of their parents. Because of the role of T-channels in determining the intrinsic firing patterns of neurons involved in absence seizures, it was suggested that these SNPs might alter channel function. The goal of the present study was to test this hypothesis by introducing these polymorphisms into a human Cav3.2a cDNA and then study alterations in channel behavior using whole-cell patch-clamp recording. Eleven SNPs altered some aspect of channel gating. Computer simulations predict that seven of the SNPs would increase firing of neurons, with three of them inducing oscillations at similar frequencies, as observed during absence seizures. Three SNPs were predicted to decrease firing. Some CAE-specific SNPs (e.g., G773D) coexist with SNPs also found in controls (R788C); therefore, the effect of these polymorphisms were studied. The R788C SNP altered activity in a manner that would also lead to enhanced burst firing of neurons. The G773D–R788C combination displayed different behavior than either single SNP. Therefore, common polymorphisms can alter the effect of CAE-specific SNPs, highlighting the importance of sequence background. These results suggest that CACNA1H is a susceptibility gene that contributes to the development of polygenic disorders characterized by thalamocortical dysrhythmia, such as CAE.
Commentary
The recent explosion of genetic studies has resulted in the discovery of an overwhelming number of new mutations associated with epilepsy. With hundreds of identified mutations reported so far, much data exist, yet there is little understanding of how altered ion channel function actually causes seizures. Some mutations severely disrupt the function or expression of important neurotransmitter receptors but cause a relatively mild phenotype (1). Conversely, comparatively subtle changes in ion channel kinetics can be associated with devastating epilepsy and mental retardation (2). With only a few exceptions, the functional characterization of these mutations has been limited to determining the expression and function of individual proteins. However, seizures are network phenomena, and dysfunction of any individual protein is only one piece of a much larger puzzle. The paper by Vitko et al. begins to bridge the gap between protein function and seizures by testing the effect of specific calcium channel variants found in patients with childhood absence epilepsy, using a computer-generated model of thalamocortical circuits.
Voltage-gated calcium channels were originally classified into three groups, named T-L– and N-type, based on the kinetic properties of the native currents (3). Subsequent cloning of the calcium channel genes has confirmed the genetic basis for this classification and also revealed the existence of multiple isoforms and splice variants for each class of calcium channel. There are three known T-type calcium channel genes, CACNA1G, CACNA1H, and CACNA1I, which encoded the Cav3.1, Cav3.2, and Cav3.3 (also known as α1G, α1H, α1I) α-subunit proteins, respectively. Like other voltage-gated calcium and sodium channels, these α-subunit proteins contain the primary pore-forming domain, which is a single peptide that is composed of four repeats of the canonical six-transmembrane domain motif (see Figure 1).
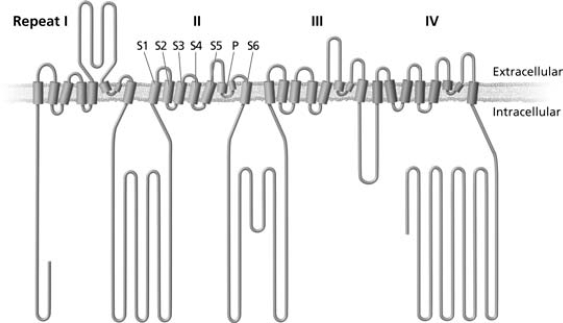
Topology of voltage-gated calcium channel α subunits. Voltage-gated calcium channels are composed of four repeated motifs, labeled in roman numerals I–IV. Each motif is composed of six-transmembrane domains (S1–S6) and a pore-forming loop (P) between S5 and S6.
Although T-type calcium channels are expressed throughout the body, they are particularly well studied in the thalamocortical circuits, where they may be involved in sleep and epilepsy. Expanding the original nomenclature, T-type calcium channels can be thought of as ticklish, transient, and tired. These ticklish channels are easily excitable at membrane potentials near the resting potential. However, inactivation also occurs at similar voltages, although the rates and extent of activation and inactivation are highly voltage dependent. At depolarized membrane potentials, activation and inactivation occur nearly simultaneously, thereby producing transient inward currents. Finally, very slow recovery from inactivation means that these quickly tiring T-type calcium channels may require hundreds of milliseconds at hyperpolarized membrane potentials before they are ready to activate again. The complex interplay of activation and inactivation endow these channels with the ability to alter neuronal activity in unusual ways. For example, during wakefulness, the T-type calcium channels in thalamic sensory relay neurons are largely inactivated, which is due to depolarizing input from the reticular activating system. However, during sleep this depolarization is lost, and the calcium channels recover from inactivation. A brief depolarization results in a transient calcium current, also known as a low-threshold spike, which may then stimulate a high-frequency burst of sodium-dependent action potentials. The presence of T-type calcium channels is a key component of the cellular machinery that allows thalamic relay cells to generate slow, rhythmic bursts of activity, even without synaptic input.
In the intact brain, these independently firing thalamic neurons project to cortex, which send excitatory feedback to the nucleus reticularis of the thalamus (nRT). The GABAergic nRT neurons then project back to the thalamic relay neurons, thereby completing the thalamocortical circuit. nRT neurons also exhibit calcium-dependent burst firing, thus allowing them to release a barrage of GABA in response to thalamocortical feedback. If the subsequent relay neuron hyperpolarization is sufficiently prolonged, previously inactivated T-type calcium channels may recover, thereby producing a low-threshold spike when the membrane potential returns to baseline. Since each nRT neuron synapses onto many thalamic relay cells, this feedback can produce synchronous burst firing among previously independently firing thalamic relay neurons. Thus, the tripartite thalamocortical circuit allows the generation of synchronous burst firing, which is the electrographic signature of slow-wave sleep.
Given the importance of T-type calcium channels in synchronizing thalamocortical circuits, it has been proposed that dysfunction of these currents could produce hypersynchronous states, such as absence seizures. This idea was supported by the discovery that one of the mechanisms of some antiabsence drugs, such as ethosuximide, may be to suppress calcium channels in thalamic neurons (4). Furthermore, studies from the Genetic Absence Epilepsy in Rats from Strasbourg (GAERS) rat model have found an age-appropriate increase in T-current amplitude in nRT neurons that was associated with increased expression of Cav3.2, but not other kinds of calcium current (5,6). Despite these encouraging animal studies, it has proven more difficult to pinpoint the role of altered calcium channels in human epilepsy.
The paper by Vitko et al. explores the role of T-type calcium channels in epilepsy. This paper builds on previous studies that found 12 different nonsynonymous single nucleotide polymorphisms (SNPs) in the CACNA1H gene from a cohort of Chinese children with childhood absence epilepsy, which were not present in control patients (7). With one exception (G773D), each SNP was found only in a single patient, and the 12 SNPs were not present in a different cohort of patients with idiopathic epilepsy (8). It is, therefore, more likely that these SNPs represent susceptibility genes, rather than major causes of absence epilepsy in the general population. As mentioned, the CACNA1H gene encodes the Cav3.2 protein, which is expressed in both cerebral cortex and nRT. In an effort to elucidate the function of these calcium channels, the authors used site-directed mutagenesis to insert the SNPs into human Cav3.2 cDNA and then expressed the proteins in immortalized HEK 293 cells. All but one of the SNPs altered the voltage-dependent kinetic properties of Cav3.2, but these subtle changes were complicated and seemingly contradictory when comparing one variant to another. However, when the altered kinetic properties were included in a computer model of thalamic relay or nRT neurons, all but two of the remaining variant Cav3.2 channels lowered the threshold for spike initiation. Furthermore, two of the variants produced spontaneous 2 to 4 Hz oscillatory firing in model nRT cells.
Childhood absence epilepsy is a polygenic disorder, thought to involve multiple genes that alter the susceptibility of an individual to develop seizures. To explore this issue further, the authors also report on several SNPs that were found in both control and childhood absence epilepsy cases. One SNP in particular (R788C) was found in four of the childhood absence epilepsy cases, including all three of the cases with the G773D variant. Analysis of the parental alleles showed that these two variants cosegregated and, therefore, were present in the same protein. Recombinant proteins containing both G773D and R788C had kinetic properties that were different from either G773D or R788C alone, resulting in an even lower threshold for firing and the ability to induce spontaneous oscillations when introduced into the model of a thalamic relay neuron.
This analysis of calcium current kinetics, while interesting, is only one step along the path to understanding the ability of ion channel mutations to cause epilepsy. For example, altered calcium influx has the potential to profoundly modify a diverse array of intracellular processes. Furthermore, it remains unknown whether these variants have normal expression and subcellular distribution in neurons. Interestingly, most of the identified polymorphisms were located outside the transmembrane domains known to be important in channel gating. Rather, the SNPs were found primarily in the cytoplasmic linker between domains I and II, raising the possibility that these channels have altered interactions with the anchoring proteins, G-proteins, or protein kinases that are known to regulate them. Additional work will be needed to explore these possibilities.
Given the complexity of these findings and the lack of an animal model, it is reasonable to remain skeptical that the calcium current kinetic changes described by Vitgo et al. are actually involved in producing absence seizures. However, the investigators’ approach to this polygenic disorder does highlight the value of considering genetic variants as epilepsy risk factors, instead of simply disease-causing mutations. More important, these findings illustrate that the complexity of neuronal physiology can make it difficult to predict the changes in the firing properties of neurons or the behavior of networks from knowledge of the biophysical properties of mutant currents. Understanding the role of ion channel mutations in epilepsy will require a deeper understanding of the multiple functions of that channel within the context of the cells and circuits in which the mutant proteins reside.