
Abstract
Despite the relatively well-characterized headache mechanisms in migraine, upstream events triggering individual attacks are poorly understood. This lack of mechanistic insight has hampered a rational approach to prophylactic drug discovery. Unlike targeted abortive and analgesic interventions, mainstream migraine prophylaxis has been largely based on serendipitous observations (e.g. propranolol) and presumed class effects (e.g. anticonvulsants). Recent studies suggest that spreading depression is the final common pathophysiological target for several established or investigational migraine prophylactic drugs. Building on these observations, spreading depression can now be explored for its predictive utility as a preclinical drug screening paradigm in migraine prophylaxis.
INTRODUCTION
Almost 70 years after Lashley's own account of visual aura implicated occipital cortex as the source (1), cortical spreading depression (SD) is now widely recognized as the neurophysiological substrate of classical migraine aura, and may be involved in migraines without a perceived aura as well. Since its discovery by Leão in the 1940s (2), SD has been a focus of intense investigation in relation to apparently diverse disease states including ischaemic or haemorrhagic stroke, head trauma and, importantly, migraine. The similarities between cortical SD and migraine aura were pointed out by Leão based on their characteristic slow spread (3), and a direct pathophysiological link has been theorized since the 1950s (4, 5). The evidence for an association between SD and migraine aura, albeit indirect, has been steadily accumulating over the past few decades with the help of technological advances.
Spreading depression
SD, observed across a broad range of species from locust to man (6–9), is a transient
depression of all spontaneous and evoked activity in neural tissue, often preceded by
a brief burst of action potential firing, which slowly propagates in brain tissue
centrifugally from the point of origin. Despite the name depression, the hallmarks of
SD are the complete and prolonged membrane depolarization of both neurons and glia,
and the associated massive K+ efflux increasing the extracellular
K+ concentration ([K+]e) from a resting level of
3–4 m
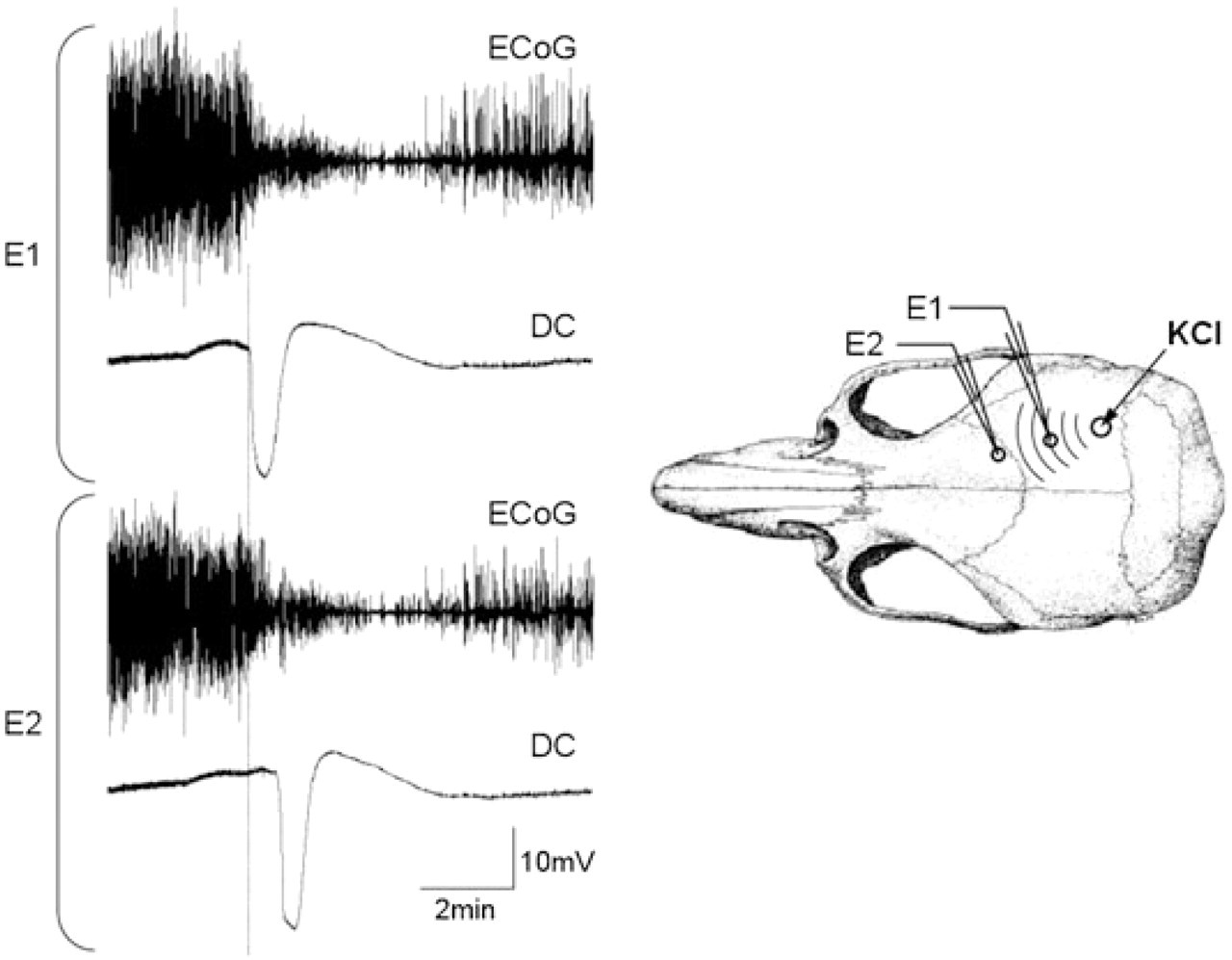
Representative tracings of electrocorticogram (ECoG) and extracellular d.c. potential during a cortical spreading depression (SD) wave in rat brain. SD is characterized by transient depression of cortical electrical activity and a slow d.c. shift 20–30 mV in amplitude lasting < 1 min. When triggered by an intense stimulus that simultaneously depolarizes a minimum critical volume of brain tissue, SD spreads centrifugally to be detected first at the proximal (E1) and, after a latency, at the distal (E2) microelectrode. In otherwise normal tissue, ECoG activity starts to recover a few minutes after the onset of SD, but may take up to 10 min to return to normal. The speed of propagation is calculated from the distance between the electrodes and the latency. SD was triggered by 1 M KCl briefly applied on to the occipital cortex, and recorded by two intracortical glass microelectrodes placed in series with the KCl application site.
SD is triggered when a minimum critical volume of brain tissue (estimated to be ∼1
mm3 in rat cortex; (12)) is simultaneously depolarized by
an intense stimulus, such as concentrated KCl application, direct electrical
stimulation, trauma, ischaemia or epileptic activity. The critical event to initiate
an SD is [K+]e exceeding a critical threshold (∼10–12
m
Although initially described in cortex, SD is known to occur readily in subcortical structures such as striatum, thalamus and hippocampus, with the exception of brainstem, which is resistant to SD unless tested in immature animals or after pharmacological preconditioning (e.g. K+ channel blockade) (19–23). Because of its dependence upon the K+ surge and to a lesser extent glutamate efflux, SD requires dense neuronal populations and limited extracellular space to propagate, and thus cannot enter white matter. Nevertheless, cortical SD can propagate into subcortical structures that have direct grey matter contiguity with the cortex (17, 24). For purposes of migraine drug screening, cortical SD susceptibility is most relevant, since cortex is most accessible, and the relationship of subcortical SD to migraine aura has not been established.
EVIDENCE SUPPORTING A ROLE FOR SPREADING DEPRESSION IN MIGRAINE
Several lines of evidence strongly, albeit indirectly, support a role for cortical SD in migraine aura:
Symptomatology of aura: Visual or somatosensory symptoms of migraine aura propagate at a rate that corresponds well to the propagation speed of experimental cortical SD, based on the known somato/retinotopic organization of human cortex (1, 4, 25). Moreover, negative symptoms of aura (e.g. expanding scotoma or hypoaesthesia) are often preceded by positive symptoms (e.g. visual scintillations or paraesthesia), resembling the brief period of neuronal burst firing preceding the suppression of activity during SD.
Cerebral haemodynamics during aura: Experimental SD is associated with a brief and variable hyperaemia lasting a few minutes, followed by an oligaemic phase lasting up to an hour during which functional coupling and hypercapnic hyperaemia are impaired. A long-lasting oligaemia similar in magnitude and duration has been detected during migraine attacks in a few studies, during which functional coupling and hypercapnic hyperaemia were diminished (26–28). A preceding hyperaemia has been more difficult to demonstrate in migraineurs (29), presumably because of the unpredictable onset of attacks and the insufficient temporal and spatial resolution of clinical imaging techniques. More recently, functional magnetic resonance imaging (MRI) during visual aura revealed a brief focal increase in brain oxygen level-dependent (BOLD) signal that propagated within the occipital cortex at a speed of ∼3 mm/min and was retinotopically congruent with the patient's visual experience (25). The BOLD signal increase was followed by a longer lasting decrease and impaired BOLD response to functional activation. These abnormalities were strictly unilateral, consistent with the unilaterality of migraine headache.
Downstream pain mechanisms: Cortical SD can activate brainstem trigeminal nucleus caudalis (30), dilate meningeal arteries with a latency fairly consistent with that of headache after aura (31), and cause plasma extravasation and inflammation (32). Furthermore, cortical SD stimulates calcitonin gene-related peptide and nitric oxide release and activates perivascular nerves (33–35).
Genetic modulation of migraine and SD: Human mutations associated with familial hemiplegic migraine type 1 (FHM1) (17, 36) and cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (Eikermann-Haerter and Ayata, unpublished data) enhance SD susceptibility in mutant mouse models. Furthermore, FHM1 mutations also augment the severity of post-SD neurological deficits, thus mimicking the phenotype in this severe human migraine syndrome (17).
Hormonal modulation of migraine and SD: Migraine is three times more prevalent in women than in men during reproductive age (i.e. menarche to menopause). Recently, SD susceptibility was shown to be significantly higher in female mice compared with male, and the sex difference was abolished by gonadectomy and ageing (17, 18).
Pharmacological modulation: Five clinically effective migraine prophylactic drugs (valproate, topiramate, propranolol, amitriptyline, methysergide) were shown to suppress SD susceptibility (37). These drugs belong to different pharmacological classes, making it unlikely that SD suppression is an incidental class effect. Furthermore, SD suppression required chronic treatment for weeks to months reminiscent of the temporal profile of their clinical efficacy in migraineurs. Interestingly, these drugs are clinically effective not only in migraine with aura but also in migraine without aura, implicating SD in the latter condition as well.
In summary, evidence strongly implicates cortical SD as the electrophysiological event underlying migraine aura, and suggests that it can trigger downstream pain pathways at least in a subset of migraineurs. Another form of spreading ionic shift, astrocytic Ca2+ waves, is unlikely to be relevant for migraine, since neither the propagation speed (≤ 1 mm/min) nor the propagation distance (< 500 μm) resembles migraine aura. Although the occurrence of SD-like propagating depolarization events has recently been shown in injured human cortex using subdural electrode strips (38), it is not yet known how cortical SD is triggered in migraineurs with aura in the absence of an underlying injury.
EVIDENCE FOR SPREADING DEPRESSION AS A SERENDIPITOUS THERAPEUTIC TARGET IN MIGRAINE
The similarities between SD and migraine aura have led a number of laboratories to test migraine drugs on SD, with mixed results. The experimental models and species used, drug doses/concentrations, administration routes and durations markedly differed, making comparisons and reconciliation difficult.
In vitro studies
Retinal SD has been a common in vitro model for pharmacological studies. Migraine drugs tested in this model, even the abortive or analgesic drugs (e.g. sumatriptan, acetylsalicylic acid), appeared to suppress retinal SD induction, duration or speed (39–41); however, the effective concentrations were often supraphysiological. Notably, only propranolol and methysergide were effective at submillimolar concentrations, perhaps suggesting that this in vitro model is relevant for migraine prophylaxis only when SD suppression is achieved at low concentrations.
In vivo studies
A total of 16 studies from eight different laboratories tested 12 prophylactic and five abortive or analgesic drugs on cortical SD after systemic administration, in vivo (Table 1). Experimental paradigms to trigger and monitor SD, and the efficacy end-points varied significantly, and all but two (37, 42) explored acute drug effects (i.e. single dose, or short-term daily treatment ≤ 1 week). In contrast to retinal SD, only three drugs, all anticonvulsant prophylactics, appeared to acutely suppress cortical SD. Tonabersat (SB-220453) suppressed KCl-evoked SDs in all four studies from one group of investigators (43–45) and their collaborators (46), whereas flunarizine showed a somewhat dose-dependent SD suppression by one laboratory (47–50), but was ineffective at a lower daily dose for 5 days in the hands of another (15). Topiramate (30 mg/kg) suppressed repetitive pinprick-induced SDs in one study when administered 30 min before testing, and the effect was reversed within 60 min (51). In contrast, a higher dose of topiramate (80 mg/kg) given as a single dose 60 min before testing, or daily for 1 week, was ineffective on SD in another study (37). Although the sources of discrepancy between the two studies are not clear, differences in the method of SD induction (pinprick vs. KCl or electrical) and monitoring (cortical blood flow and single unit activity vs. d.c. potential) may be responsible. Apart from the three drugs, acute in vivo studies did not support the overall notion that SD is a target in migraine prophylaxis.
Systemic administration of selected migraine drugs on cortical spreading depression (SD), in vivo

Dur, SD duration; Amp, SD amplitude; ND, not determined or reported; DC, slow extracellular potential shift; LDF, laser Doppler flowmetry measuring blood flow changes; SUA, single unit spike activity; E, electrically-induced SD; M, mechanical pinprick-induced SD; KCl, KCl-induced SD.
A number of potential caveats could explain the lack of efficacy of prophylactic
drugs on SD, in vivo, such as anaesthetic influences, differences in
tissue properties (e.g. no blood flow in retina preparation), and the sensitivity of
techniques used to trigger and monitor SD. It has also been suggested that a
predisposition to SD (e.g. injury) might be required for migraine drugs to suppress
SD (47). More
importantly, however, higher tissue drug concentrations may have been attained
in vitro, since brain levels upon systemic administration would
depend on pharmacokinetic factors (e.g. plasma levels and half-life) as well as drug
penetration across the blood–brain barrier. In support of this, topical application
of serotonin (5-HT) reuptake inhibitors citalopram (2.5–12.5 m
Chronic treatment (weeks to months) is required for some prophylactic drugs to suppress SD, in vivo
In all the negative studies with classical prophylactics reviewed above, drugs have been administered either as a single dose or as daily doses for up to a week. However, it is well recognized that migraine prophylactic drugs show a gradual build up of clinical efficacy over a few months, and a possible need for chronic treatment to achieve efficacy on experimental SD has been proposed (47). Indeed, recent data strongly suggest that SD suppression is a mechanism of action shared by several migraine prophylactic drugs after prolonged treatment. Five established prophylactic drugs (valproate, topiramate, propranolol, amitriptyline and methysergide) are now shown to suppress cortical SD susceptibility after daily treatment for 4 weeks or more in rats (37). All five drugs elevated the electrical threshold for SD induction, reduced the frequency of SDs triggered by topical KCl application (Fig. 2), and decreased the SD propagation speed in a dose-dependent manner, without altering SD duration or amplitude. More importantly, longer treatment durations (up to 4 months) produced more potent SD suppression, reminiscent of the need for prolonged treatment with these drugs in migraineurs to reduce attack frequency and/or severity (59, 60). Consistent with most previous reports, single doses of these prophylactic drugs did not alter SD susceptibility or electrophysiological properties when administered 60 min before testing. D-propranolol, deemed clinically ineffective (61, 62), did not suppress SD susceptibility even after prolonged treatment, perhaps demonstrating the specificity and predictive value of the models used in this study.

Representative tracings of extracellular d.c. potential during continuous topical KCl application for 2 h, recorded from two rats treated with intraperitoneal injections of saline or valproate (200 mg kg−1 day−1) for 4 weeks. Seventeen cortical spreading depressions (SDs) are detected at the proximal recording site (E1) in the saline-treated rat; only nine SDs reached the distal electrode (E2), suggesting propagation block between the two electrodes. The number of SDs detected at E1 was nine in the valproate-treated rat, and only two propagated to E2. This experimental model (i.e. frequency of continuous KCl-induced SDs) shows good correlation with electrical threshold of cortical SD (see Fig. 3). The electrode configuration was identical to that shown in Fig. 1.
It is important to note that not too many drugs have been tested as rigorously in SD susceptibility models, and independent confirmation by other laboratories will help build confidence in the predictive ability of these models. Nevertheless, these data suggest that prolonged treatment is a requisite for some migraine prophylactic drugs to suppress SD. Even with drugs that do suppress SD after brief treatment, longer treatment durations may augment their efficacy, as shown for fluoxetine (42). The reasons for the delayed appearance of efficacy on SD are not clear. As noted above, for some drugs it may simply be a matter of achieving therapeutic brain tissue concentrations, in vivo; those drugs are then expected to suppress SD via the same mechanisms when tested at higher concentrations in vitro, or topically in vivo. However, ≥ 4 weeks' treatment may be too long for tissue levels to reach a plateau, and alternative mechanisms should be considered for some drugs. It is known that prolonged treatment with some migraine prophylactic drugs alters gene expression (63). Detailed discussion of individual gene products altered by each drug is beyond the scope of this review, except to note that many of the altered genes are mechanistically related to SD such as those involved in glutamatergic neurotransmission (e.g. tissue glutamate levels), brain water homeostasis (e.g. aquaporin-4), neurotransmitter release, K+ channels and gap junctions (64, 65). Therefore, drugs that require weeks to suppress SD susceptibility may do so not directly by interfering with receptors, ion channels or pumps, but by altering gene expression. A potential consequence of altered gene expression, one that may require even longer treatment durations, is a change in tissue morphology or ultrastructure, such as synaptic or mitochondrial density, and even glia/neuron ratios. Such possibilities remain hypothetical at this time, although differences in cortical thickness have been reported in migraineurs (66). Drugs acting exclusively by altering gene expression or ultrastructure after prolonged prophylactic therapy would not be expected to suppress SD acutely even when tested at higher concentrations in vitro, or topically in vivo.
PHARMACOLOGICAL TARGETS MODULATING SPREADING DEPRESSION SUSCEPTIBILITY AND THEIR RELEVANCE IN MIGRAINE PROPHYLAXIS
Migraine drugs that have been shown to suppress SD belong to different pharmacological and therapeutic classes (e.g. anticonvulsant, β-adrenergic blocker, antidepressant, and serotonergic), suggesting that their proximate pharmacological effects converge onto SD. Multiple mechanisms of action have been defined for most migraine prophylactic drugs, and a review of their numerous pharmacological targets exceeds the scope of this review. However, it is important to consider some of the potential drug targets that suppress SD, as well as relevant others that have been ineffective.
Glutamate receptors
Both competitive (e.g. DL-2-amino-7-phosphonoheptanoic acid) and non-competitive NMDA receptor blockers (e.g. ketamine, phencyclidine, MK-801, CP-101,606) are potent inhibitors of SD; they dose-dependently elevate electrical SD threshold, slow down or block SD propagation, and shorten SD duration (56, 67–76). None of the clinically efficacious migraine drugs (prophylactics in particular) appear to interact ‘directly’ with the NMDA receptor, but may suppress excitatory neurotransmitter release (77). It is worth noting, however, that ketamine and the inhalational analgesic nitrous oxide, both of which inhibit NMDA receptors (78) and suppress SD (56, 70, 71, 79, 80), reportedly show clinical efficacy in aborting migraine aura (81) and migraine headache (82, 83), respectively. NMDA receptor blockers have a number of side-effects that have thus far limited their usefulness in neurotherapeutics, and their role in migraine prophylaxis depends on the development of agents that are clinically better tolerated (e.g. glycine site blockers, NR2B inhibitors). Mg2+ augments voltage-dependent block of NMDA receptors, and suppresses SD susceptibility in vitro (8, 84–86). Mg2+ showed both abortive and prophylactic efficacy in some but not all trials (87–89). Acute intravenous administration of Mg2+ reportedly suppressed KCl-induced SDs in the rat (90); however, whether chronic Mg2+ supplementation can elevate brain tissue levels to have an impact on SD is unknown.
Unlike NMDA antagonists, inhibitors of non-NMDA subtypes of ionotropic glutamate receptors have generally been ineffective on SD (67, 91–96), except when SD was triggered by agonists of these receptors or by high-frequency afferent stimulation (94, 95, 97). For this reason, topiramate-induced SD suppression is unlikely to be related to its inhibition of non-NMDA receptor-mediated neurotransmission (98, 99). It should be noted, however, that it is not known how SD is triggered in migrainous human brain, and it may in part involve intense synaptic activity to elevate [K+]e above the SD threshold. If so, inhibitors of non-NMDA subtypes of ionotropic glutamate receptors may still suppress SD in migraineurs.
Some prophylactic drugs may interfere with glutamatergic neurotransmission indirectly by decreasing glutamate release (e.g. amitriptyline, gabapentin). One such drug is lamotrigine, which may be more efficacious on migraine with aura (100–102), but has not been tested on SD.
Voltage-gated Ca2+ channels
Non-selective inhibition of voltage-gated Ca2+ channels (e.g. by divalent cations Co2+, Ni2+, Mn2+) suppresses SD (16, 47, 95, 103, 104). The relevant channel subtypes appear to be the P/Q- and N-types (95, 105), although in two studies inhibitors of these channels failed to suppress pinprick-induced SD (105, 106). The importance of P/Q-type voltage-gated Ca2+ channels in SD susceptibility is further underscored by modulation of SD susceptibility in mouse models expressing mutations in this channel (17, 36, 107). Because P/Q and N-types are the major channels mediating presynaptic Ca2+ influx, the mechanism of SD suppression may relate to inhibition of glutamate release (107, 108). Among the drugs with clinical efficacy in migraine, only gabapentin has been shown to interact directly with P/Q-type channels and reduce glutamate release as a major mode of action (109). Whether gabapentin can suppress experimental SD has not yet been tested. Flunarizine, which shows efficacy in SD suppression as well as in migraine prophylaxis, is a relatively non-selective inhibitor of voltage-gated Ca2+ channels (110–112). Topiramate also inhibits L-, R- and P/Q-type channels (113–115). Valproate inhibits T-type channels, but there is no evidence at this time to support a role for T-type channels in SD. Both lamotrigine and levetiracetam reportedly suppress N- and P/Q-type Ca2+ currents in cortical neurons; however, their efficacy in migraine is not well established (100, 116, 117), and it is not known whether they suppress SD.
Adrenergic receptors
Because of the known efficacy of some β-adrenergic receptor blockers in preventing
migraine, a few studies have tested the role of adrenergic receptors on SD. Both
noradrenaline (0.1–1 m
Propranolol is a non-selective β-adrenergic receptor blocker with no intrinsic sympathomimetic activity; β-adrenergic receptor blockers that possess partial agonistic activity are ineffective in migraine prophylaxis (118). Propranolol also has potent membrane stabilizing actions; however, β-blockers without this action appear to be equally effective clinically. As mentioned above, propranolol suppresses SD both in vitro and in vivo. Interestingly, chronic treatment with amitriptyline decreases β-adrenergic receptor expression (63), which may be relevant in SD suppression after prolonged administration of this drug in rats (37).
The mechanism of adrenergic modulation of SD may relate to modulation of presynaptic glutamate release. Activation of β-adrenergic receptors augments glutamatergic neurotransmission via increased presynaptic cyclic adenosine-3′,5′-monophosphate levels, and enhanced Ca2+ influx possibly through P/Q-type Ca2+ channels (119–122). Consistent with this, propranolol inhibits the stimulatory effect of β-adrenergic receptor activation on evoked glutamate release, in vitro (120, 123). Conversely, α2-adrenergic receptors inhibit depolarization-induced glutamate release (124–126), and may explain SD suppression by clonidine.
Serotonin receptors
5-HT receptors also modulate SD susceptibility. Migraine attacks have been associated
with a relatively 5-HT-depleted state, and reserpine, a drug that depletes 5-HT in
the brain, can acutely precipitate migraine attacks, although long-term use reduces
the attack frequency (127, 128). Consistent with this, 5-HT depletion by tryptophan hydroxylase
inhibitor para-chlorophenylalanine augments SD susceptibility (129), while 5-HT reuptake inhibitors
amitriptyline,
Although in general 5-HT2 receptors stimulate and 5-HT1A receptors inhibit neuronal excitation, there are data suggesting that the more relevant receptor subtype in SD suppression is the 5-HT1A receptor. For example, 5-HT1A receptor agonist 8-OH-DPAT reduced SD duration in rat cortical slices (67). Topical application of yohimbine, an α2-antagonist and 5-HT1A agonist, suppressed CSD in the rat in vivo, and this effect was blocked by 5-HT1A antagonist NAN-190 (55). NAN-190 also increased the frequency of SDs triggered by topical KCl, suggesting that the endogenous 5-HT exerts an inhibitory effect on SD via this receptor. The mechanisms of SD suppression by 5-HT1A receptors may relate to the activation of G-protein-coupled K+ channels and hyperpolarization, inhibition of adenylate cyclase, and to reduced glutamate release possibly via inhibition of N- and P/Q-type Ca2+ channels (67, 132–137).
Gap junction blockers
Although, based on indirect evidence, a role for neuronal synchronization via gap
junctions at the onset of SD has been proposed (138), it has been difficult to
ascertain the role of astrocytic or neuronal gap junctions in SD, mainly due to the
lack of selective inhibitors. In retina and rat hippocampal and neocortical slices,
non-selective gap junction blockers (e.g. aliphatic alcohols) block SD propagation at
millimolar concentrations (96, 139–142). However, more recent data
using the more selective gap junction inhibitor carbenoxolone (100 m
K+ channels
In general, an increase in membrane K+ conductance decreases neuronal excitability by virtue of hyperpolarization. Consistent with this, blockade of K+ channels (e.g. tetraethylammonium, 4-aminopyridine, Ba2+, Cs+) has been shown to augment SD susceptibility and even trigger spontaneous SDs (151–154). Conversely, suppression of repetitive KCl-evoked SDs and their propagation speed has been shown acutely after systemic treatment with a K+ channel (Kv7) opener with good brain penetration (155). In the absence of studies with selective inhibitors or activators, it is unclear whether these effects are specific for particular channel subtypes or a non-specific consequence of membrane hyperpolarization. Nevertheless, K+ channels may be a target for some migraine prophylactic drugs (e.g. valproate).
Sigma-1 receptors
Sigma-1 receptors (σ1R) are unique non-opioid, non-phencyclidine receptors that reside in the endoplasmic reticulum and are mainly expressed in the central nervous system (156). They modulate intracellular Ca2+ signalling, K+ channels, glutamatergic transmission, and neurotransmitter release and neuronal excitability in a complex manner (157). Several σ1R agonists have been shown to suppress KCl-induced and hypoxic SD, in vitro, and this effect appeared to be independent of NMDA receptors (158). As with the K+ channels, future in vivo work with more selective σ1R agonists and antagonists will help clarify the potential role of this receptor in SD suppression, and whether currently available migraine prophylactic drugs that are known to interact with this receptor (e.g. fluoxetine, fluvoxamine) suppress SD by this mechanism.
Tissue pH
Low pH, whether caused by direct acidification, or indirectly by carbonic anhydrase inhibition or hypercapnia, is a potent suppressor of neuronal excitability in general, and SD susceptibility in particular (152, 159–164). Although the efficacy of carbonic anhydrase inhibitor acetazolamide in migraine is unclear (116, 165), there is anecdotal evidence for its benefit in hyperexcitable channelopathies such as FHM (166–168). Topiramate also inhibits carbonic anhydrase and causes metabolic acidosis, a well-recognized side-effect, which may contribute to its SD suppression and migraine prophylactic efficacy (169, 170). Although no correlation between arterial blood pH and the degree of SD suppression was found after chronic topiramate treatment in rats (37), brain tissue pH is more relevant and was not measured in this study.
Na+/K+ ATPase
Because SD initiation requires elevation of [K+]e above a
critical threshold (∼10–12 m
GABAA receptors
Drugs that augment GABAergic transmission, despite being potent anticonvulsants, do
not inhibit SD. For example, diazepam did not alter electrical SD threshold and
increased propagation speed and duration (56). Similarly, clonazepam did not
inhibit pinprick-induced SD propagation in the cat monitored by laser Doppler
flowmetry and single unit activity recordings (52). Barbiturates also did not
suppress SD, in vitro or in vivo (16). For example,
diethylbarbiturate (0.8 m
Voltage-gated Na+ channels
It is generally agreed that voltage-gated Na+ channels, while involved in the process, are not critical for SD (94, 184–188). One exception, of course, is when SD is evoked by high-frequency afferent stimulation requiring action potential firing. For example, topical tetrodotoxin superfusion increased the electrical stimulation (20 Hz) threshold of SD, but did not block SD initiation by KCl or its propagation once initiated (189). Conflicting data have recently been reported using pinprick to trigger cortical SD after systemic tetrodotoxin administration, suggesting model differences in sensitivity to pharmacological modulation (106). Inhibition of voltage-gated Na+ channels may still be a relevant mechanism of action by some migraine prophylactic drugs (e.g. topiramate, valproate, amitriptyline, and fluoxetine), if intense synaptic activity is a requisite for SD to develop in migraineurs. Recent identification of Na+ channel mutations in FHM3 provides further support for a potential role for these channels in SD initiation (190).
Non-pharmacological interventions
Transcranial magnetic stimulation (TMS) has recently attracted attention as a potential migraine therapy. TMS decreased headache intensity when delivered shortly after attack onset (191), but although prophylactic TMS anecdotally reduced attack frequency and intensity (192), a larger, controlled, blinded trial showed no difference from placebo in primary and secondary end-points (193). TMS does alter cortical excitability, and its experimental equivalents in rodents have been shown to modulate SD (194–196). In a recent preliminary report, TMS administered shortly before or immediately after cortical pinprick appeared to acutely suppress SD susceptibility monitored using surrogate blood flow changes (197). However, both the experimental and clinical data are too preliminary to draw conclusions on the utility of TMS to suppress SD or migraine.
In summary, a multitude of direct and indirect pharmacological effects has been described for migraine therapeutics, and there appears to be no single unifying mechanism to explain or predict their efficacy in SD suppression. To complicate the matter further, mechanisms of action of drugs that require prolonged treatment for weeks to months may differ from those that acutely suppress SD after a single dose, underscoring the need to develop experimental SD susceptibility models for high-throughput drug screening.
EXPERIMENTAL MODELS OF SPREADING DEPRESSION FOR DRUG SCREENING
Since its discovery and initial characterization by Leão in anaesthetized rabbits, numerous SD models have been described, each with its own merits and shortcomings. A detailed account of technical aspects of SD models can be found in the volume by Bures et al. (16). Quantitative assessment of SD susceptibility for the purpose of drug screening requires consideration of some practical aspects of experimental models to improve intra- and interobserver reproducibility, as well as the sensitivity and specificity (i.e. predictive value) of the assays.
In vitro vs. in vivo models
In vitro, SD has been studied in retina, brain slices and neuronal cultures. Much of the SD work was done using amphibian and later chick retina models; chick retina forms a natural slice without a need for cutting, with a few well-characterized neuronal layers and no blood vessels. Subsequent in vitro work largely relied upon cortical, hippocampal and cerebellar slices. The advantages of in vitro SD models include bypassing the blood–brain barrier by drug superfusion, and lack of systemic physiological, pharmacokinetic and anaesthetic confounders, allowing high-throughput standardized drug testing as has been done in retina in a few studies. The disadvantages include exposure to trauma and hypoxia during tissue preparation and, more importantly, the difficulty in estimating clinically relevant drug concentrations in vitro. Because of these caveats, pharmacological SD suppression in vitro has not always been predictive of in vivo efficacy.
In vivo SD models have been described in many species from pigeons to cats and monkeys, and there is considerable interspecies variability in SD susceptibility. A great deal of variability also exists among the models used, particularly in the methods used to trigger and detect SD, route of drug administration, type of anaesthetic, and the extent of systemic physiological monitoring. Although most brain regions can sustain SD, in vivo drug testing has been carried out almost exclusively in cerebral cortex. In vivo assays of drug efficacy are expected to be more predictive of clinical efficacy (except when drugs are topically applied on the cortex). However, systemic physiological state, which has so often been neglected, has a significant impact on SD susceptibility. Most importantly, body temperature, plasma glucose, arterial blood gases and pressure, and possibly pH, are important determinants of SD occurrence, duration, refractory period and propagation speed, and need to be routinely monitored throughout the experiments. Drugs with systemic physiological or cerebrovascular effects may indirectly alter SD properties, sometimes without even crossing the blood–brain barrier. For example, hypotension and hypertension may have been the cause of prolonged and shortened SD durations by clonidine and sumatriptan, respectively (30, 47). Blood glucose can be standardized across groups by overnight fasting, which tends to facilitate SD. Anaesthesia is a potential confounder, in vivo. Although certain commonly used anaesthetics can directly impact SD susceptibility, there is no evidence at this time to suggest that the choice of anaesthesia alters the efficacy of a drug to suppress SD susceptibility, except when the anaesthetic and the drug are known to act on the same target (e.g. barbiturate anaesthesia when testing a GABAA receptor agonist, or ketamine anaesthesia when testing an NMDA receptor antagonist). In general, it is advisable to avoid anaesthetics that strongly suppress SD susceptibility, such as ketamine. While barbiturates, urethane and α-chloralose do not significantly suppress SD, inhalational anaesthetics and nitrous oxide have mild but reproducible inhibitory effects (80).
Methods to trigger SD
Both in vitro and in vivo, the most common and reliable methods to trigger an SD have been topical or intraparenchymal application of KCl, electrical stimulation, and trauma. Although ultimately they all evoke SD by elevating the [K+]e above the critical threshold, the way this is achieved by each stimulus type is highly relevant for drug efficacy.
KCl can be used to assess SD susceptibility in several ways. For example, KCl concentration threshold to trigger an SD (as a surrogate for [K+]e threshold) can be determined by applying escalating concentrations of KCl topically, or intraparenchymally via microdialysis or iontophoresis (107). Alternatively, escalating volumes of KCl solution at a fixed concentration can be applied by intraparenchymal microinjection (18), which probably is also a measure of the minimum critical volume of depolarized tissue to trigger SD. A third approach is to determine the frequency of repetitive SDs triggered by continuous topical application of KCl at a fixed concentration (17, 37, 80); SD frequency increases in a KCl concentration-dependent manner in this model. This method not only measures SD susceptibility, but is also sensitive to the absolute refractory period after an SD (16), which is about 2–3 min depending on the physiological state of the tissue.
Direct electrical stimulation using cathodal currents may be a better method to determine actual SD threshold, but requires careful and reproducible control of stimulus geometry, which is a function of electrode shape and impedance characteristics as well as cortical surface properties (13, 16, 198). The stimulus can be delivered by monopolar or bipolar needle, or cortical cup electrodes; the latter provides better control over stimulus geometry, but all methods give thresholds between 10 and 103 μC, and can be used as long as the electrode configuration and impedance are consistent when comparing groups. Both single square pulses (typically 102–104 ms, 10–103 μA) and high-frequency stimulus trains have been used to evoke SD locally at the stimulation site. Alternatively, a high-frequency stimulus can be delivered to an afferent axonal pathway to evoke SD by intense synaptic activity (i.e. mimicking seizure activity). It is important to note that the mechanisms of [K+]e elevation, and therefore the pharmacological sensitivity profiles, may differ between a single prolonged square pulse and high-frequency tetanic stimulation. For example, the latter is likely to require action potentials and synaptic transmission, and may be suppressed by inhibitors of voltage-gated Na+ channels or non-NMDA type ionotropic glutamate receptors. For the same reason, glutamate receptor agonists (e.g. NMDA), which can evoke SD when applied directly onto the tissue (199), are not suitable to trigger SD for drug screening as they yield an obviously different pharmacological sensitivity profile. Because the mechanisms of SD initiation in human brain are not known, the experimental SD induction method most predictive of clinical prophylactic drug efficacy is not clear.
Trauma, usually in the form of pinprick or gentle mechanical pressure onto the tissue, is also a common method to evoke SD. However, unlike the KCl and electrical stimulation, it cannot be easily graded to determine SD thresholds, and may be insensitive to detect small changes in SD susceptibility. The suppression of pinprick-induced SD is often an all-or-none response in a given animal, quantified as percent of animals in which SD is blocked (51). Moreover, cumulative injury may prohibit repetitive SD induction using this method, further limiting its statistical power. In some studies, pinprick-induced SD has shown a decidedly different pharmacological profile compared with KCl-induced SD, in that it was effectively suppressed by inhibitors of voltage-gated Na+ channels (and by an inhibitor of non-NMDA subtype of ionotropic glutamate receptors and an agonist of GABAA receptor in a preliminary report), but not by inhibitors of N- or P/Q-type voltage-gated Ca2+ channels (105, 106, 200), further underscoring the importance of identifying the most relevant SD model for drug screening.
Lastly, ischaemia is a potent trigger for SD. For example, endothelin has been used to evoke SD, probably due to vasoconstrictive ischaemia (201). A role for transient ischaemic events as triggers for migraine aura in a subset of patients is further supported by the association between migraine with aura and intracardiac or pulmonary right-to-left shunts (e.g. patent foramen ovale) (202). Paradoxical microembolism as a migraine trigger is a plausible hypothesis yet to be tested, and further work is required to develop experimental models to assess ischaemic SD susceptibility.
Methods to monitor SD
The standard method to detect SD and study its electrophysiological properties is extracellular d.c. potential recordings. This is best accomplished by two or more intraparenchymal glass micropipettes placed serially along the path of propagation, but surface recordings using metal electrodes also suffice, albeit with less precision. The SD attributes most often measured in this way are amplitude and duration (at half maximal amplitude) of the d.c. potential shift, as well as the propagation speed or failure between the two recoding sites. The depression of synaptic activity (e.g. electrocorticogram, or single unit spike activity) can supplement but should not substitute for d.c. potential recording, as they can be suppressed by drugs that have no impact on SD itself.
Alternative methods to detect SD, such as intrinsic optical signal changes reflecting intracellular/extracellular volume shifts, in vitro, or cerebral blood flow changes, in vivo, can supplement the d.c. potential recordings. However, they should not substitute for d.c. potential recording to detect SD, since their mechanisms are poorly understood, and at least in case of cerebral blood flow changes, they can be uncoupled from electrophysiological measures of SD (203). Finally, MRI can be used to detect SD reliably (46, 204, 205), but this method will probably be cost-effective only in larger gyrencephalic animals.
EMPIRICAL CONSIDERATIONS
Developing experimental drug screening models in migraine prophylaxis requires validation, i.e. demonstration of predictive value of the model for clinical efficacy. Ideally in a given model, all drugs known to be effective in migraine prophylaxis should suppress SD, and those tested and found to be ineffective should not. Congruence, however, may be incomplete for several reasons, including differences between experimental animals and humans (e.g. intrinsic brain factors, drug pharmacokinetics), sensitivity and specificity of the employed experimental models, and importantly, the quality of clinical trials of migraine prophylaxis. On the latter point, only a handful of drugs have been conclusively tested for their clinical efficacy in migraine prophylaxis, and lack of efficacy has been particularly difficult to ascertain (118, 206). All these underscore the need to characterize and validate relevant and predictive preclinical SD models and attributes to use as end-points for drug development in migraine. Some broad outlines of a drug screening paradigm can be empirically drawn to seek validation of the models initially, and then to test candidate drugs. Far from having been validated, however, the outlines below are intended to be recommendations rather than guidelines, and further work will undoubtedly help highlight some models among others towards that goal.
Experimental models
Despite the advantages of in vitro models mentioned above, drug screening ultimately needs to demonstrate efficacy in vivo. Because different types of stimuli to evoke an SD have yielded different pharmacological profiles for its suppression, it is critical to identify the method that is most relevant for clinical efficacy. In our experience, quantitative assessment of SD susceptibility, a poorly defined term in itself, is best achieved using KCl or electrical stimulation, because they both allow precise titration of stimulus intensity, and because SD susceptibility measured using these stimuli was sensitive to suppression by clinically effective migraine prophylactic drugs, suggesting good predictive value (37). Regardless of the trigger, however, it is generally agreed that SD propagating into adjacent regions acquires a rather uniform character, provided that the stimulus intensity is comparable. Hence, the best method to monitor SD is less ambiguous, since the characteristic d.c. potential shift accompanied by depression of cortical activity is historically the gold standard. Last but not least, careful choice of anaesthesia and systemic physiological monitoring are critical for proper data interpretation.
Relevant SD attributes
The most direct attribute of SD susceptibility is probably electrical or KCl
threshold for triggering an SD. One has to keep in mind that the experimental models
detecting SD at a distance assess not only SD initiation but also successful spread
to the recording site. Because the ionic shifts sustaining the momentum of spread may
be weaker or more prone to inhibition than the external stimulus used to initiate the
SD, drugs may preferentially slow down and block propagation rather than initiation
of SD. Based on our own experience, this is often the case especially when
suprathreshold stimuli are used (e.g. 1
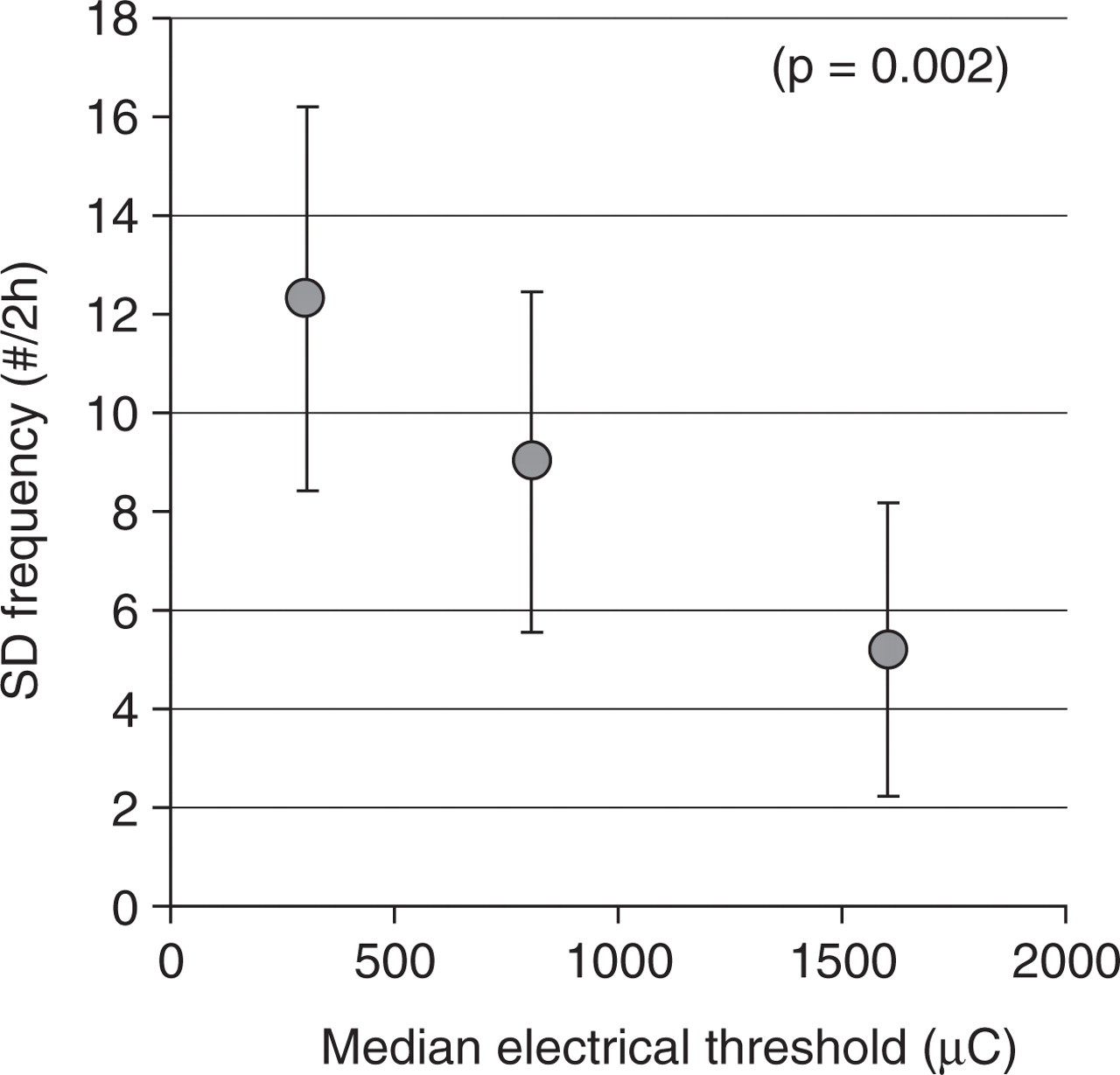
Graph showing the inverse relationship between the electrical threshold and the
frequency of spreading depressions (SDs) evoked by continuous topical KCl
application, when these two were measured in the same animal
(n = 65 rats). The data are pooled vehicle and drug
treatment groups from Ayata et al. (37). Vertical axis, mean
±
Experimental animals
SD susceptibility varies among species (e.g. lissencephalic brains are generally more susceptible than gyrencephalic brains), often attributed to glia/neuron ratios (164, 207). Moreover, it is not known whether potential drug targets (e.g. gap junctions, voltage-gated Ca2+ channels, NMDA receptors) contribute to SD occurrence and propagation to the same extent in different species. Therefore, initial high-throughput studies in more cost-effective and accessible rodents should ideally be followed by demonstration of efficacy in gyrencephalic species (e.g. cat). In addition, some drugs may show efficacy or lose their efficacy in the presence of certain genetic risk factors (e.g. P/Q-type Ca2+ channel inhibitors in FHM1 with mutations in this channel); it might therefore be of interest to test drug candidates in genetic mouse models of migraine as well, as they become available. Because migraine shows a female preponderance, it will be important to screen drugs in both sexes.
Treatment dose and protocol
In general, drug doses used in rodents are approximately an order of magnitude higher than those used in humans, mainly due to faster rates of metabolism and elimination. However, it is difficult to predict the clinically relevant animal doses for SD suppression for a given drug. Measurement of plasma levels may help guide dose selection, when this information is available from human studies. Further complicating the matter, brain tissue levels may not always correlate with plasma levels, and may show gradual build-up upon prolonged treatment as a requisite for efficacy in migraine prophylaxis. Even when target plasma levels are achieved, prolonged treatment may be necessary for efficacy to become manifest, possibly via altered gene expression and/or ultrastructural changes. Therefore, a thorough drug screening should seek a dose–response relationship, and start by testing the acute effects of a single administration, followed by chronic treatment in search of increased efficacy and/or reduced dose requirements.
CONCLUDING REMARKS
Although there is little doubt that SD is the electrophysiological substrate of migraine aura, whether it is the inciting event for migraine headache is as yet unresolved. If SD is indeed the trigger for migraine headache, SD inhibitors will probably be useful in prophylaxis rather than abortive treatment, because the brief and episodic nature of SD (i.e. aura) is unlikely to provide a wide enough window of opportunity to intervene pharmacologically with the process before pain mechanisms are activated. Exceptionally, if an attack can be anticipated (e.g. exposure to known triggers, hormonal fluctuations), then a drug that acutely suppresses SD after a single or few doses can be used as a ‘pre-emptive abortive’ therapy (i.e. episodic rather than chronic treatment). On a more speculative note, prophylactic suppression of SD susceptibility is expected to reduce migraine attack frequency by preventing SD occurrence. In addition, headache severity might be reduced by limiting the duration or distance of propagation of breakthrough cortical SDs (i.e. propagation block), although there is yet no evidence that these attributes correlate with headache intensity.
It is possible that multiple yet unidentified genetic factors, modulated by hormonal and environmental influences, determine SD susceptibility and response to different prophylactic drugs in migraineurs. Identification of biomarkers for SD susceptibility (e.g. electrophysiological, genetic) that have better sensitivity and specificity than symptomatic aura might therefore help predict the response to anti-SD therapy and tailor patient selection.
Since the earlier attempts exploring SD as a target, it has taken more than two decades to develop the concept and put it to use prospectively as a drug-screening paradigm for migraine. Only a handful of prophylactic drugs have been vigorously tested in relevant SD models. Tonabersat may be considered the first migraine drug candidate that is being tested in clinical trials, in part based on its efficacy to suppress experimental SD. Although its precise mechanism of action is unknown and clinical efficacy remains to be proven, the momentum needs to be carried forward by screening new drugs and candidates in standardized SD models. It is equally important, however, to test drugs that are conclusively ineffective in migraine prophylaxis to select and refine relevant SD models and optimize their predictive value. In this respect, the quality of clinical trial design, conduct, analysis and reporting will be critical (206).