
Abstract
Migraine is a common, recurrent, primary headache disorder associated with significant morbidity as well as high direct and indirect costs. Despite its impact, only a proportion of migraineurs who meet criteria for prophylactic treatment take preventive medication. Antiepileptic drugs and β-blockers are among the most used preventive therapies, but their exact mechanisms of action in migraine prophylaxis are unknown. Recent research has pointed to the role of cortical spreading depression in the genesis of migraine aura and pain, with neuronalglial gap junctions playing a prominent part in cortical spreading depression. Tonabersat is a unique compound with demonstrated activity as a gap-junction inhibitor in animal studies. In preclinical and clinical trials, tonabersat was well tolerated, with no cardiovascular effects; the pharmacokinetic profile suggested its usefulness in the prophylaxis of migraine.
Introduction
Migraine is a recurrent primary headache disorder ranked 19th by the World Health Organization among diseases that cause disability (1). Typical features are unilateral location, pulsating quality, moderate or severe pain, aggravation by or avoidance of routine physical activity, nausea, photophobia and phonophobia (1). Migraine occurs with or without aura, the focal, transient neurological symptoms that precede or accompany pain. Headache, nausea or photophobia generally begins immediately after or within 1 h of aura (1, 2). Some patients may experience a premonitory phase hours or days before onset of migraine symptoms or a resolution phase, or both (1).
Burden of migraine
Epidemiology
The 1-year prevalence of migraine rises between infancy and approximately 40 years, peaks between ages 35 and 45 years and then declines (2). Migraine is less prevalent in Africa and Asia than in Europe, Central and South America and North America (2). Women with migraine outnumber men by a ratio of 2 : 3 (2).
A comparison of studies conducted between 1989 and 2004 in the USA demonstrated that migraine prevalence remained stable throughout that time (3). In 2004, a survey of 162 576 individuals ≥ 12 years of age revealed an 11.7% 1-year prevalence (17.1% in women, 5.6% in men), with a peak in midlife and a lower frequency among adolescents and persons > 60 years old. Among migraineurs, 31.3% reported three or more episodes per month, and 53.7% were severely impaired or required bed rest. Moreover, 25.7% of migraineurs met criteria for prophylaxis, and a further 13.1% were considered potential candidates for preventive treatment. Only 13.0%, however, took daily preventive medication (3).
Direct and indirect costs
According to population-based survey data, two-thirds of the financial burden of migraine is attributable to indirect costs such as inability to perform household duties and absence from work (4). In 2004, direct costs accounted for $5.21 billion in out-patient care, $4.61 billion in prescriptions, $0.73 billion in in-patient care and $0.52 billion in emergency department care—for a projected national migraine burden of $11.07 billion (5). In another study conducted from 2002 to 2003, indirect costs to employers, most due to absenteeism, were approximately $12 billion (6).
Migraine has a high prevalence and significant direct and indirect costs. There is a large population that could benefit from a new effective prophylactic treatment to reduce the impact of migraine.
Mechanisms
The vascular theory of migraine, proposed in the 1940s, postulated that migraine aura resulted from cerebral vasoconstriction and migraine headache was secondary to cerebral vasodilation (7). By the early 1980s, it was shown that migraine aura was associated with oligaemia spreading anteriorly over 15–45 min and that severe headache could be present without hyperaemia. A theory based only on vasoconstriction and vasodilation, therefore, cannot fully explain the phenomena of headache in migraine (8).
It now appears likely that migraine headache is associated with sensitized nociceptors that send increasingly intense stimuli (peripheral sensitization resulting from neuroinflammation of the dural and meningeal nociceptors) to the trigeminal nucleus caudalis (TNC). This results in central sensitization, which is associated with abnormal neuronal excitability in the TNC, and amplification of pain (abnormal processing) (9). Thus, migraine-specific triggers may promote primary brain dysfunction that causes activation of the sensory fibres of the trigeminal nerve (10).
Cortical spreading depression
Cortical spreading depression (CSD) has been implicated in the mechanism of migraine aura (1). In CSD, a wave of electrical activity moves across the cerebral cortex at a rate of 2–3 mm/min. CSD is associated with neuronal excitation and then depression. During CSD, shifts in cortical steady-state potential cause transient increases in levels of calcium, potassium, nitric oxide (NO) and glutamate, accompanied by brief increases, followed by sustained decreases, in cerebral blood flow (11–13). Satellite glial cells (astrocytes) that surround the neuronal cell body may mediate this wave of activity (14).
Astrocytes control the neuronal microenvironment and modulate neuronal function. Neurons and glial cells communicate by way of gap junctions—channels formed by connexins that allow ions and other small molecules to pass from one cell to another—and modulate the excitability of both cell types (14). In experimental studies, activation of neuronal–glial communication in one region of the trigeminal ganglion led to activation in other ganglionic regions (14). Activation of one trigeminal region through gap junctions thus may lower the sensory threshold in other trigeminal nerve afferents, thereby expanding the sensory inputs that get processed centrally. Activation of the trigeminal neurons leads to release of calcitonin gene-related peptide. This 37-amino-acid regulatory neuropeptide is widely expressed in peripheral and central neurons and may be responsible for pain transmission (10).
The relationship between CSD and migraine pain remains to be completely clarified (15). CSD may be responsible for both aura symptoms and, by stimulating local nociceptors, migraine headache (11). CSD releases potassium, arachidonic acid and NO, which could all activate trigeminal afferents. Trigeminal activation can result in vasodilation and plasma leakage within the dura mater (Fig. 1) (15, 16). Spreading depression in the cerebellum or in silent areas of the cortex may underlie migraine without aura, and parasympathetic nerves controlled by the brainstem may also activate trigeminal neurons (13).
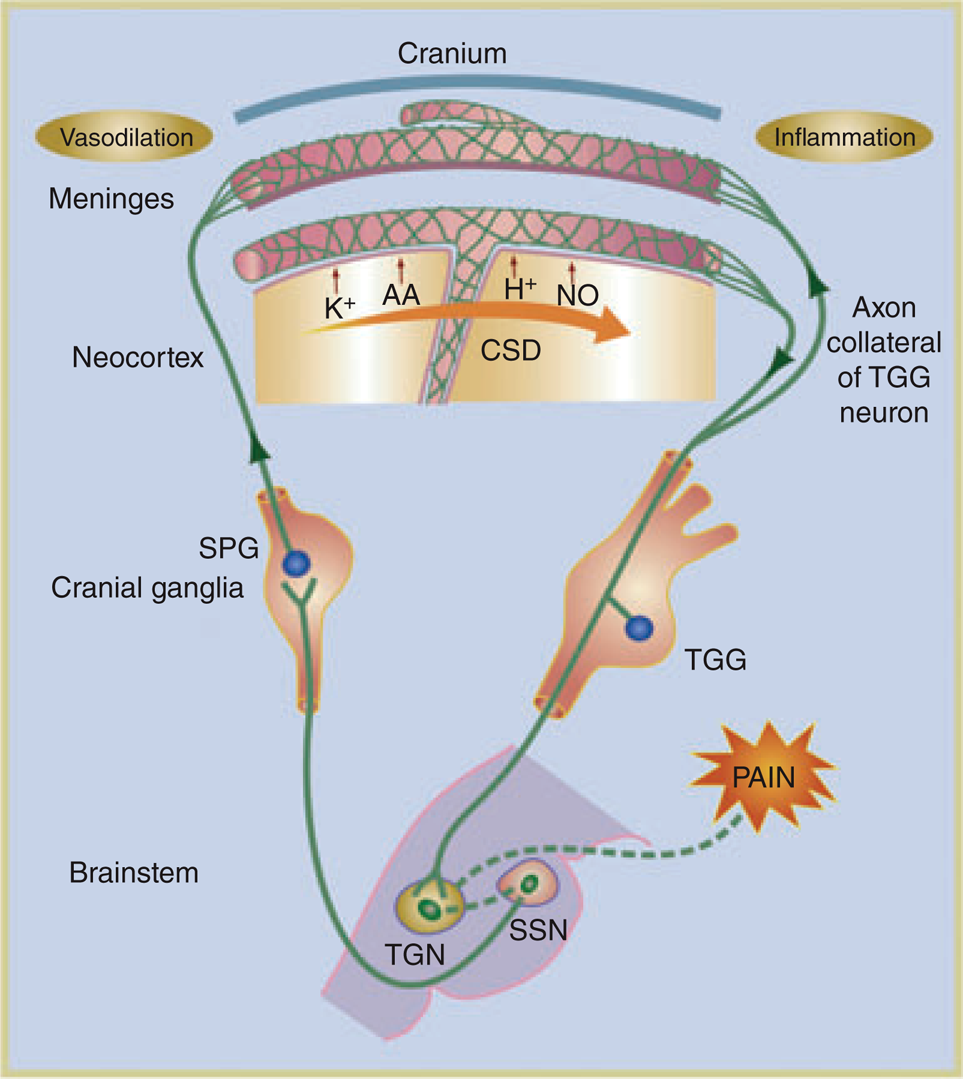
The relationship among trigeminal nerve, meninges, brainstem, and parasympathetic efferents. Cortical spreading depression (CSD) results in the release of H+, K+, arachidonic acid (AA) and nitric oxide (NO). These agents diffuse towards local blood vessels and depolarize perivascular trigeminal terminals, which, in turn, activates the caudal portion of the trigeminal nucleus (TGN) in the brainstem. Collateral axons of activated neurons in the trigeminal ganglion (TGG) release proinflammatory peptides in the meninges and their vessels, leading to a local inflammatory reaction. The activation of TGN caused by CSD produces vasodilation of meningeal vessels. The perception of pain is mediated by higher-order projections from the TGN. Adapted from Iadecola (16).
Prophylaxis of migraine
Evidence-based consensus guidelines, published by the American Academy of Neurology, articulate several goals of migraine treatment: (i) reduction in frequency and severity of attacks and migraine-associated disability; (ii) reduction of patients' reliance on poorly tolerated or ineffective acute pharmacotherapies; (iii) improvement in quality of life; and (iv) avoidance of escalating use of acute medications (17). Table 1 lists the goals of migraine prevention.
Consensus guidelines for preventive migraine therapy

Adapted from Silberstein (17).
Migraine-preventive agents can lower healthcare costs, as shown by a recent retrospective analysis of resource utilization in a large claims database. The addition of preventive medication reduced use of drugs for acute attacks and decreased visits to doctors' surgeries and emergency departments (18).
Most drugs used for migraine prophylaxis in the USA lack that specific indication. A review of clinical studies supported the use of eight drugs available in the USA for migraine prevention—topiramate, divalproex sodium/sodium valproate, amitriptyline, metoprolol, propranolol, timolol and extracts of butterbur—as well as two non-pharmacological approaches, relaxation therapy and biofeedback (19). This review discusses only topiramate and propranolol, both indicated for migraine prophylaxis (20, 21).
Topiramate
Topiramate and other antiepileptic drugs target multiple cortical and subcortical loci, altering voltage-gated ion channels and chemical transmission to decrease abnormal brain excitability (22).
Precisely how topiramate exerts its anticonvulsant and migraine prophylactic effects is unknown; however, preclinical studies have identified four properties that may account for the drug's efficacy in epilepsy and migraine prophylaxis: (i) blockage of voltage-dependent sodium channels; (ii) augmentation of the activity of the neurotransmitter γ-aminobutyric acid (GABA) at some subtypes of the GABA-A receptor; (iii) antagonism of the AMPA/kainate subtype of the glutamate receptor; and (iv) inhibition of the carbonic anhydrase enzymes, particularly isozymes II and IV (20). These actions together appear to enhance GABA-mediated inhibition and to decrease glutamate- and calcium-mediated excitation, two complementary activities that may act in concert to prevent a migraine attack (23).
Topiramate is effective in the prophylaxis of migraine, reducing the number of attacks by ≥ 50% compared with placebo (24). The most common adverse events (AEs) are fatigue, paraesthesia, dizziness, hypoaesthesia, nausea, diarrhoea, abdominal pain, dyspepsia, difficulty with memory and with concentration/attention, insomnia, anxiety and mood problems (20). Decreasing rates of these events during an open-label extension study suggest tolerability over long-term use (25, 26).
Propranolol
Propranolol caused reductions of 44% based on headache diaries and 65% based on clinical improvement ratings and global patient reports in large meta-analyses. Fatigue and loss of energy or desire (27) are frequent AEs. The mechanism of action of propranolol in prophylaxis of migraine is unknown (21).
Tonabersat: potential for migraine prophylaxis
The proposed mechanism of migraine suggests that modulation of gap junctions may be useful in migraine prevention. Tonabersat (SB-220453), a novel benzoylamino benzopyran compound (28), inhibits CSD and neurogenic inflammation, induces carotid dilation and decreases cortical NO concentrations during CSD (29–32). Tonabersat acts uniquely at a stereo-specific binding site that could be associated with the neuronal/glial gap junction (33). None of over 100 pharmacological agents, including triptans and valproate, demonstrated affinity at this site (32). In animal studies, tonabersat had no cardiovascular effects and lacked significant central nervous system (CNS) adverse effects, even at very high doses (28–32).
Clinical trials of tonabersat
The efficacy and safety of a single 15, 20, 25, 40 or 80-mg dose of tonabersat taken acutely at the onset of migraine with or without aura were examined in three multicentre, randomized, double-blind, placebo-controlled, parallel-group trials. Two studies, one international and one North American, were dose-ranging studies in which the efficacy and safety of tonabersat 15, 40 or 80 mg and 25, 40 or 80 mg, respectively, were examined (34). The third study was an international study in which the efficacy and safety of tonabersat 20 and 40 mg, and sumatriptan were examined (35). Review of the total intent-to-treat population (n = 1400) showed tonabersat to be safe and well tolerated.
Efficacy
The primary efficacy measure in all three trials was headache relief 2 h post treatment (34, 35). Secondary parameters in the international and North American dose-ranging studies were headache relief (after 1 and 4 h) or abolition (after 1, 2 and 4 h); recurrence within 24 h; time to recurrence; use of rescue medication 2 h after treatment; and time to meaningful relief (34). In the second international study, the secondary efficacy variables were headache relief (after 1 and 4 h), abolition of headache (1, 2 and 4 h), need for additional migraine medication within 60 min of the 2-h post-treatment assessment and meaningful relief 2 h post treatment (35).
The efficacy outcomes differed among the studies (34, 35). In the international dose-ranging study, significantly more patients in the tonabersat groups reported headache relief after 2 h compared with the placebo group. In the North American dose-ranging and second international studies, tonabersat 20 and 40 mg provided no clinically or statistically significant advantage over placebo in relieving acute migraine (Table 2).
Headache relief 2 h post treatment

∗Statistically significant, P = 0.013.
†Statistically significant, P = 0.002.
‡Not statistically significant vs. placebo.
§Not compared statistically against placebo or tonabersat.
In the international dose-ranging study, 37% of the tonabersat 15-mg group and 41% of the 40-mg group reported relief 2 h postdose, compared with 21% of the placebo group (P = 0.013 and P = 0.002, respectively) (34). A bell-shaped dose–response curve was evident for headache relief at 2 h, however. Tonabersat was more efficacious than placebo in relieving moderate or severe headache at 2 and 4 h and in abolishing headache at 4 h. The reasons for the low efficacy with the 80-mg dose and the difference in efficacy between the trials are unclear.
Previous sumatriptan use had an impact on the efficacy of tonabersat (34, 35). Patients who had not previously taken sumatriptan had a better response to tonabersat. A large difference in prior sumatriptan exposure distinguished the population of the higher-efficacy study (international dose-ranging) from patients in the other two studies. In the international dose-ranging study, approximately 25% of patients had used sumatriptan. In the North American dose-ranging study, by contrast, almost 66% of the enrolment noted sumatriptan use, as did 47% of the second international study population.
Prior sumatriptan exposure was not thought to be relevant initially and hence was not tested as a covariate in the statistical models. The response rate, however, was approximately twice as high among sumatriptan-naive patients in both the 25-mg (22.39%, previous sumatriptan; 43.75%, no sumatriptan) and 80-mg (24.62% and 47.62%) tonabersat groups (34).
A treatment-centre effect emerged in the international dose-ranging study, which was conducted at 35 sites in Australia, Belgium, France, Germany, South Africa and the UK (34). Headache relief 4 h after dosing revealed a treatment-by-centre interaction. Patients at centres in France (54% of the total) had a higher placebo response than did those at non-French centres. A significantly higher proportion of patients at non-French centres receiving tonabersat 80 mg had relief at 4 h than did those receiving placebo (69% vs. 45%) (P = 0.009), but at French centres, none of the tonabersat doses was significantly more efficacious than placebo. On further analysis, the differences were not country specific but, rather, French vs. non-French centres. No further explanation for this variance has yet emerged.
Absorption profiles in these studies were less than ideal for rapid onset of action. In healthy individuals, the time to maximum plasma concentration (T max) ranged from 0.5 to 3 h; however, a Phase 2A study that used tonabersat during an attack identified a median T max of 3 h (range 1.5–14 h), presumably due to delay of gastric emptying, which has been reported in migraineurs during an attack.
Safety
In the three studies of tonabersat, the overall incidence of AEs that were suspected or probably related to study medication was 13–33% for all treatment groups and 13–23% with placebo. In the North American dose-ranging study, 13, 23 and 33% of patients who received tonabersat 25, 40 and 80 mg, respectively, and 23% of patients given placebo reported AEs (34). In the international dose-ranging study, 16, 27 and 20% of patients who received tonabersat 15, 40 and 80 mg, respectively, and 13% of patients given placebo reported AEs. In the second international study, 21 and 32% of patients receiving tonabersat 20 and 40 mg, respectively, 20% of patients taking sumatriptan and 14% of patients given placebo reported AEs (35).
Among AEs that were suspected or considered to be probably related to treatment across all three studies, dizziness, nausea, vertigo, somnolence, paraesthesia, abdominal pain and palpitation were most common (occurring in at least 3% of any group) (34, 35). The incidence of these events rose with increasing doses in each study, with the exception of nausea, somnolence and palpitation in the international dose-ranging study, which had a higher incidence at 40 mg than 80 mg. Nausea, dizziness and vertigo generally appeared within 2 h of dosing and in the 2–24 h thereafter, with a lower likelihood after 2 h. The 2-h onset suggests fairly rapid CNS penetration.
Few cardiovascular AEs were reported in any of the tonabersat groups. In the North American dose-ranging study, a prolonged QT interval in one patient was judged not likely to be related to study medication; a T-wave inversion in another patient had a suspected relationship to tonabersat (34). Both changes occurred 9 days after drug ingestion. In the other two studies, no abnormal electrocardiogram results at follow-up were considered of significance to study medication (34, 35). Two cardiovascular AEs in the second international study were considered severe: peripheral ischaemia starting 30 min after administration of tonabersat 40 mg and lasting 3 h and palpitations with sumatriptan, probably unrelated to the drug (34). No serious AEs or deaths considered related to tonabersat occurred, and no patients withdrew owing to AEs (34, 35). Tonabersat had no clinically significant effect on vital signs, electrocardiogram tracings or laboratory values.
Glyceryl trinitrate-induced experimental migraine model
The pharmacological activity and pharmacokinetic properties of tonabersat suggested promise in the prophylaxis of migraine. The short-term preventive potential of tonabersat was investigated in a study that used a glyceryl trinitrate (GTN) infusion (0.5 µg/kg) to induce migraine on two occasions 60 min after patients were given tonabersat 40 mg or placebo by mouth following an overnight semifast (36). Of the 15 entrants, nine received GTN on both days; seven patients completed the study (two patients withdrew after the first GTN-provoked headache) (36). Data comprised self-reported migraine and migraine that fulfilled International Headache Society criteria for all comparisons (36). A comparison of tonabersat and placebo was possible in nine patients, none of whom reported a reduction in immediate headache after taking tonabersat (mean 3.0 vs. 3.4, median 3 vs. 3) (P = 0.55). Delayed peak headache after 60 min but before 12 h numerically was lower after tonabersat (mean 3.8 vs. 5.0, median 4 vs. 5) (P = 0.11), as was overall peak headache between drug administration and 12 h (mean 4.6 vs. 5.8, median 4 vs. 7) (P = 0.15), but headache score was not significantly numerically reduced (36).
Although preclinical screening of tonabersat did not detect vascular effects, the study was discontinued due to an apparent synergism between GTN and tonabersat that resulted in potentially serious hypotension, which in two cases produced serious AEs. Thus, the number of patients studied was too small to achieve statistically significant results (36). In this model, tonabersat did not appear to have preemptive efficacy compared with placebo; however, these results do not apply to the drug's effects in long-term prophylaxis of spontaneously occurring migraine (36).
Tonabersat in migraine prophylaxis
A pilot Phase 2A multinational, randomized, double-blind study evaluated tonabersat for the prophylaxis of migraine in 123 patients (n = 65 placebo, n = 58 tonabersat) (37). On entry, patients experienced between 4 and 14 migraine days per month (within 2–6 attacks with or without aura). After a 4-week baseline, patients were assigned in a 1 : 1 ratio to receive tonabersat or placebo. Tonabersat was dosed at 20 mg/day for 2 weeks and then 40 mg/day for 10 weeks, with safety follow-up 7 days post treatment. The primary end-point was mean change from baseline to month 3 in monthly migraine days. Compared with placebo, tonabersat reduced the mean monthly number of migraine days (−4.4 days vs. −3.7 days), although this was not statistically significant (P = 0.14). Among other measures, tonabersat compared with placebo produced a higher proportion of patients with a ≥ 50% reduction in the number of migraine days (59% vs. 49%, P = 0.22) and attack frequency (62% vs. 45%, P < 0.05) during month 3 relative to baseline (Fig. 2) and decreased the number of days on which rescue medication was taken during month 3 relative to baseline (−3.7 days vs. −2.0 days, P = 0.02). AEs reported in a higher percentage of patients in the tonabersat group than in the placebo group were nausea (17% vs. 6%), dizziness (9% vs. 5%), vertigo (9% vs. 0%) and headache (7% vs. 2%). AEs affected 51% of the placebo group and 61% of the tonabersat group. One patient withdrew from the placebo group because of memory impairment, hypoaesthesia and dizziness; two patients left the tonabersat group because of drowsiness, dizziness and nausea.

The percentage of patients with a ≥ 50% reduction in the number of migraine days and migraine attacks during the third month of treatment relative to baseline. ∗P < 0.05 vs. placebo. Adapted from Goadsby et al. (37).
The potential for migraine prophylaxis with tonabersat 40 mg and 80 mg once daily compared with placebo is being evaluated in 500 patients in an ongoing Phase 2B, randomized, double-blind, 20-week trial. The primary end-point is the reduction in mean monthly attacks during the last 8 weeks of treatment, determined by patients' entries on electronic diary cards. An open-label, long-term extension is also in progress.
Conclusion
CSD is a well-validated model for a mechanism underlying migraine aura and may be one generator of headache pain (12). Experimental evidence suggests that CSD is propagated, at least in part, by neuronal gap junctions (12, 13).
In animal models of migraine, tonabersat appears to act at the neuronal gap junction (33), inhibiting CSD, reducing neurogenic inflammation and decreasing NO concentrations during CSD (29–32). Tonabersat exerted no cardiovascular effects and was without significant CNS effects, even at high doses.
In humans, the pharmacokinetic profile of tonabersat is favourable with respect to its potential use in migraine prophylaxis. Median T max occurred between 0.5 and 3 h after administration of 2- to 80-mg doses. The average terminal half-life of 24–40 h also suggests long-lasting protective effects after achievement of steady state.
Results from Phase 2 prevention trials, a unique mechanism of action and pharmacokinetic and safety profiles suggest the potential value of tonabersat in migraine prophylaxis. Ongoing studies are evaluating the efficacy of tonabersat in the prophylaxis of migraine.
Footnotes
Competing interests
S.D.S. is a paid consultant to Minster Pharmaceuticals and owns stock in the company.
Acknowledgements
A. Harkavy (RBH Associates, Inc., Elmhurst, NY, USA), and P. Kontur, PhD and M. Kersting, PhD (JL Shapiro Associates Inc., Edison, NJ, USA) assisted with manuscript preparation with financial support from Minster Pharmaceuticals.