
Abstract
Two identical randomized, placebo-controlled, crossover studies were conducted to evaluate consistency of response to sumatriptan/naproxen sodium 85/500 mg (S/NS) over four attacks in adults with migraine. Patients were instructed to treat within 1 h of pain onset while pain was mild. Co-primary end-points were pain-free response at 2 h (2hPF) and 24-h sustained pain-free response (24hSPF) calculated as percentages of all attacks. In Study 1, 570 patients treated 1693 attacks with S/NS and 424 with placebo. In Study 2, 565 patients treated 1678 attacks with S/NS and 422 with placebo. Compared with placebo, S/NS conferred higher 2hPF rates (Study 1: S/NS 52%, placebo 25%; Study 2: S/NS 50%, placebo 20%; both
Introduction
Migraine is a chronic disorder with episodic attacks (CDEAs) of neurological dysfunction. Like other CDEAs, migraine is characterized by recurrent, often debilitating attacks and an enduring predisposition to attacks (1). Although attack rates vary from person to person, many patients experience frequent migraine episodes. In a large population-based study, 63% of respondents meeting International Classification of Headache Disorders, 2nd edn (ICHD-II) criteria for migraine with or without aura reported one to four migraine attacks per month (2). In a study of 100 patients with migraine seeking care at a headache centre during November and December 2005, the monthly frequency of headaches was three to five in 53% of patients and at least six in 33% of patients (3).
Given the chronic, recurrent nature of migraine, acute medications are utilized to treat many attacks over a long period of time. This pattern of use could impact efficacy and/or tolerability of medication. In addition, most patients identify consistent efficacy over multiple migraine attacks as a desirable attribute of acute migraine therapies (4). It is therefore important with any acute migraine therapy to assess consistency of efficacy and tolerability over multiple migraine attacks. A double-blind, multiple-attack, crossover design with random insertion of placebo facilitates the assessment of consistency of response both within and among subjects (5). Variants of the crossover design with interspersed placebo, used in previous studies with sumatriptan (6–8) and rizatriptan (9, 10), eliminates the drawbacks of more common, less rigorous approaches—namely, the long-term, uncontrolled, open-label safety study (11–13) and the parallel-group, placebo-controlled, multiple-attack study (14–16). The open-label safety study lacks a placebo control group and is often subject to participation bias as responders selectively enrol (5). The placebo-controlled, multiple-attack study is associated with high attrition in the placebo group and attack-to-attack carryover effects arising from use of the same treatment for all attacks (5).
This paper described the results of two randomized, double-blind, placebo-controlled, crossover studies designed to evaluate the consistency of response to the combination tablet sumatriptan/naproxen sodium 85/500 mg (S/NS) in the treatment of multiple migraine attacks. S/NS was developed based on the clinical observation of enhanced efficacy with combination therapy and with the physiological rationale of targeting multiple putative mechanisms involved in the pathogenesis of migraine (17). S/NS has been shown to be more effective than either of its components in replicate clinical trials utilizing factorial designs (17). Data from a 12-month, open-label study suggest that response is maintained across multiple attacks (18), but response consistency has not previously been assessed in randomized trials.
In the studies described here, patients were asked to practise ‘early intervention’ by treating migraine attacks within 1 h of their onset and while pain was mild. In contrast, the other placebo-controlled, crossover studies assessing response consistency asked patients to wait for moderate or severe pain before treating (6–9). This approach of early intervention while pain is mild is widely advocated because it has been associated with higher pain-free rates at 2 h and higher sustained pain-free rates at 24 h (19–25). The two multiple-attack crossover studies reported here compared S/NS with placebo administered for the treatment of migraine while pain was mild.
Methods
Patients
Men and non-pregnant, non-lactating women were eligible for the studies if they were 18–65 years old, had at least a 6-month history of migraine with or without aura as defined by the ICHD-II (26), had an average of two to six migraine episodes monthly during the 3 months preceding the screening visit, typically experienced moderate to severe migraine pain preceded by an identifiable mild pain phase, and could distinguish between mild migraine pain and that of other headache types. Women had to be physiologically incapable of becoming pregnant or, if they could become pregnant, to agree to practise adequate contraception during the study.
Patients were excluded if they had more than six migraine attacks or < 15 headache-free days per month in any of the 3 months before screening; uncontrolled hypertension (sitting systolic pressure ≥ 140 mmHg, sitting diastolic pressure ≥ 90 mmHg); confirmed or suspected cardiovascular or cerebrovascular disease, ischaemic abdominal syndromes, peripheral vascular disease, or Raynaud's syndrome; a history of cardiac arrhythmias requiring medication or clinically significant electrocardiographic abnormalities that, in the investigator's opinion, contraindicated study participation; or basilar or hemiplegic migraine. Other exclusion criteria included use within 3 months before screening of migraine prophylactic medications containing ergotamine, ergot derivatives, or methysergide; use of a monoamine oxidase inhibitor within 2 weeks or preparations containing St John's Wort within 4 weeks before screening; current use of any anticoagulant or antiplatelet agent (except aspirin ≤ 325 mg/day for cardiovascular prophylaxis); and use of a migraine prophylactic regimen that had not been stable for at least 2 months prior to screening. Patients were eligible regardless of whether they were triptan or naproxen naive. All patients provided written informed consent prior to study participation.
Procedures
The protocols for these identically designed, randomized, double-blind, placebo-controlled, crossover studies (GlaxoSmithKline protocols TRX103632 and TRX103635) were approved by ethics committees or institutional review boards for each of the 105 US study sites. At the screening visit, patients meeting eligibility criteria were randomized to one of the following five treatment sequences for the treatment of four migraine attacks at home (P = placebo; A = active treatment: S/NS 85/500 mg): PAAA, APAA, AAPA, AAAP, or AAAA. The AAAA treatment sequence, in which patients received S/NS in each period of the crossover, was included for comparison with treatment sequences with interspersed placebo in order to assess carryover effects and within-subject consistency. Study medication was a fixed-dose, single-tablet formulation containing sumatriptan succinate equivalent to sumatriptan 85 mg and naproxen sodium 500 mg. The study medication was delivered in a fast-disintegrating, rapid-release formulation designed to facilitate tablet disintegration and drug dispersion and to mitigate the effects of gastric stasis that can accompany migraine (25, 27). Patients were instructed to practise early intervention in treating migraine attacks during the study—that is, to use study medication within 1 h of onset of migraine head pain while the pain remained mild. Patients returned for clinic visits 4–7 days after the first treated migraine attack as well as 4–10 days after treatment of the fourth migraine attack (or upon premature withdrawal from the study) or 4 months after screening visit, whichever occurred first.
No barbiturates or barbiturate-containing compounds, opioids or opioid-containing compounds, ergotamine-containing compounds or ergot derivatives, or 5HT1 agonists could be taken within 24 h before or after treatment with study medication. Non-steroidal anti-inflammatory drugs (except daily aspirin ≤ 325 mg/day for cardiovascular prophylaxis), anti-emetics and other headache medications were prohibited from 24 h before to 2 h after dosing with study medication; all other analgesics (e.g. paracetamol) were prohibited from 6 h before to 2 h after dosing with study medication. Monoamine oxidase inhibitors were prohibited throughout the study. Patients were permitted to take rescue medication beginning 2 h postdose as prescribed or recommended by the physician. The recommended rescue medication regimen was two naproxen sodium 220-mg tablets and, if needed, one additional 220-mg tablet 6 h later, not to exceed three 220-mg tablets in 24 h. A second dose of study medication was not permitted.
Measures
Efficacy data were summarized from patient diary records of headache pain (none, mild, moderate, severe); assessments of nausea, vomiting, photophobia and phonophobia (present/absent); and use of rescue medication predose and at designated postdose time points through up to 24 h for each treated attack. Efficacy data were analysed for the intent-to-treat population, defined as randomized patients who treated at least one attack and provided an evaluation of randomized treatment. The co-primary efficacy end-points were the proportion of attacks that became pain free (i.e. post-treatment pain severity of 0 with no use of rescue medication) 2 h after treatment across attacks and the percentage of attacks with sustained pain-free response (i.e. no pain from 2 to 24 h postdose with no use of rescue medication) across attacks. The use of two co-primary end-points, one assessing early post-treatment efficacy [2-h pain-free response (2hPF)] and the other sustained benefits [24-h sustained pain-free response (24hSPF)] provides a broader and more stringent test of consistency of response than in previous multiple-attack studies.
Data analysis
The primary end-point was analysed using generalized estimating equations including terms for treatment sequence, treatment and period (i.e. time). The appropriateness of the model was tested using terms for carryover effects and interaction. Differences between S/NS and placebo were tested separately for each of the co-primary end-points using a step-down procedure (28) to control the type 1 error rate; adjusted
Secondary across-attack efficacy measures included the percentages of attacks with (i) migraine-free response (i.e. no pain, nausea, vomiting, photophobia or phonophobia and no use of rescue medication) 2 and 4 h postdose and (ii) presence of photophobia, phonophobia and nausea (considered separately) 2 h postdose. Secondary efficacy measures calculated for each attack separately included the percentages of patients with (i) 2hPF; (ii) sustained pain-free response; (iii) migraine-free response 2 and 4 h postdose; (iv) presence of photophobia, phonophobia and nausea (considered separately) 2 h postdose; and (v) use of rescue medication. (Other secondary efficacy measures will be reported in another manuscript.) Across-attack analyses of secondary end-points were performed using generalized estimating equations including terms for treatment sequence, treatment and period (i.e. time). Differences between S/NS and placebo were tested separately for secondary across-attack efficacy end-points using a closed, sequential approach (i.e. hierarchical step-down method) to control the overall type 1 error rate. By-attack data were analysed with the Cochrane–Mantel–Haenszel test controlled for baseline values. Analyses of post-baseline symptoms (nausea, vomiting, phonophobia, phonophobia) were controlled for the presence/absence of the symptom at baseline. Unlike the analyses for other secondary end-points, that for rescue medication use was not adjusted for multiple comparisons.
Within-subject measures of response consistency included (i) 2hPF postdose and (ii) sustained pain-free response in at least one of two, two of three, and three of three attacks treated with S/NS. To assess within-patient consistency and possible operation of carryover effects, these within-subject efficacy measures were computed both for consecutive attacks treated with and without S/NS with an additional interspersed placebo-treated attack. These measures were summarized with descriptive statistics.
Tolerability was assessed by calculating the incidence of specific adverse events, defined as any untoward medical occurrences regardless of their suspected cause, that were reported by a patient or noted by a clinician during the study. Adverse event data were summarized with descriptive statistics for the safety population, defined as randomized patients who treated at least one attack. The overall incidence of adverse events was calculated based on occurrences per treated attack. In addition, the percentages of patients with individual adverse events reported in ≥ 2% of patients by attack within 72 h of treatment with study medication, with adverse events leading to premature withdrawal from the study, and with serious adverse events (defined as adverse events that resulted in death, disability, or incapacity; were life threatening; required or prolonged hospitalization; or were a congenital anomaly or birth defect) were calculated.
Results
Patients
Figure 1 summarizes the disposition of patients in each study. The numbers of patients randomized to treatment were 646 in Study 1 and 620 in Study 2. Safety analyses included the 570 patients in Study 1 and 565 patients in Study 2 who were randomized and treated at least one migraine attack. Efficacy analyses included the 568 patients in Study 1 and 563 patients in Study 2 who took study medication and had at least one post-baseline efficacy evaluation.

Patient disposition.
In Study 1, 570 patients treated 1693 attacks with S/NS and 424 attacks with placebo. In Study 2, 565 patients treated 1678 attacks with S/NS and 422 attacks with placebo. In Study 1, the numbers of patients treating attacks 1, 2, 3 and 4 with S/NS were 447, 429, 418 and 399, respectively. In Study 2, the numbers of patients treating attacks 1, 2, 3 and 4 with S/NS were 458, 423, 402 and 395, respectively. Demographics and baseline clinical characteristics were similar between studies (Table 1).
Demographics and baseline clinical characteristics
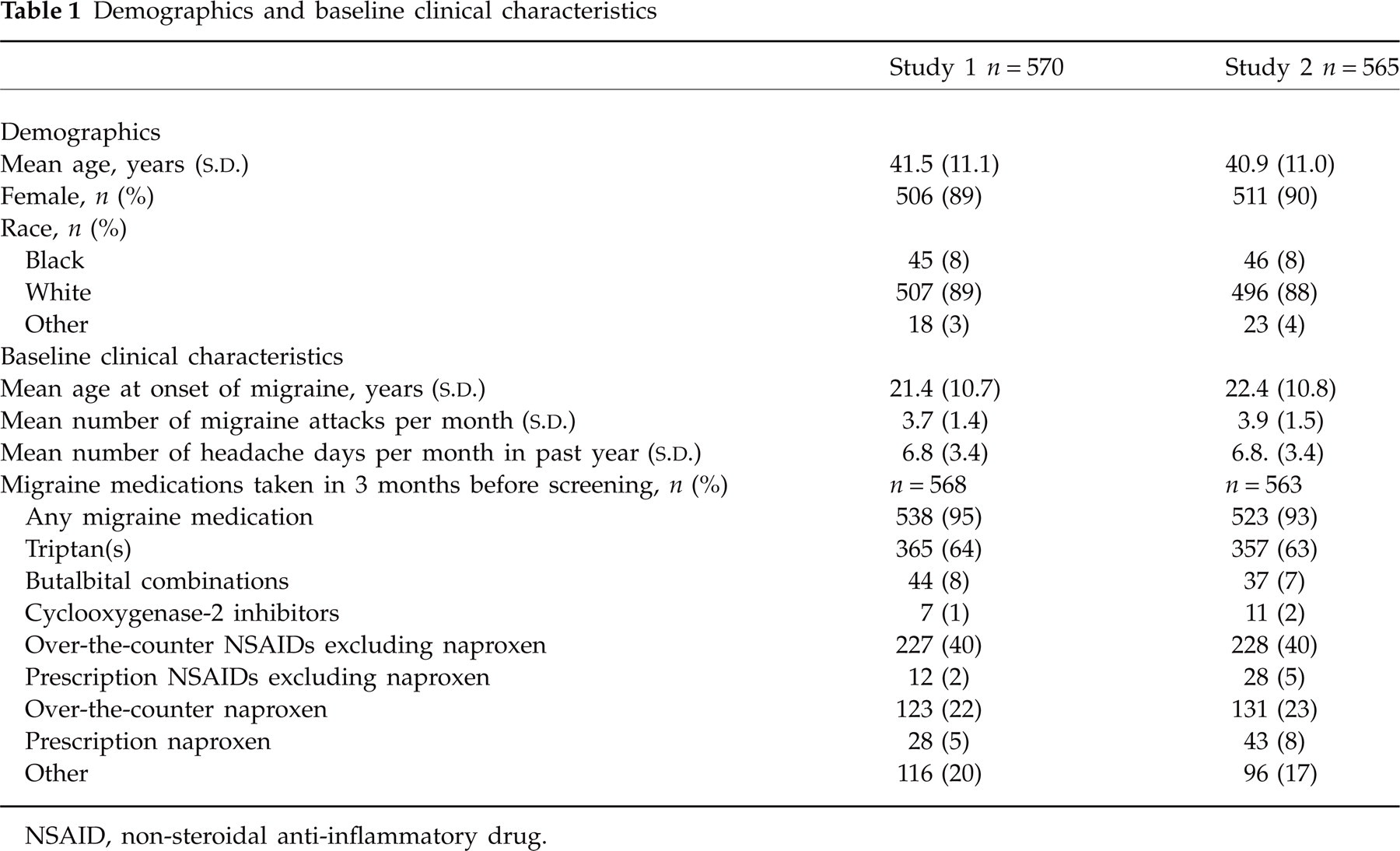
NSAID, non-steroidal anti-inflammatory drug.
Summary measures of efficacy
Pain-free response 2 h postdose
Across attacks in both studies, 2hPF postdose (a co-primary end-point) was reported in more attacks after treatment with S/NS than placebo [Study 1: S/NS 52%, placebo 25%; difference 28%; 95% confidence interval (CI) 21, 36;
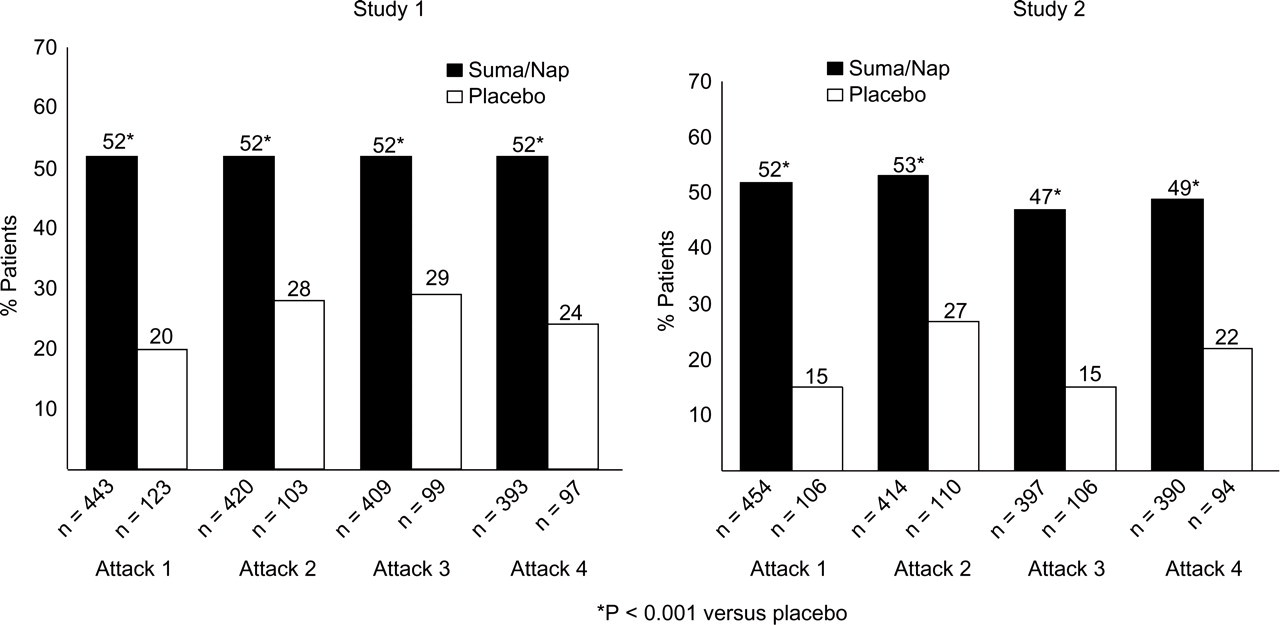
Pain-free response 2 h after dosing with sumatriptan/naproxen (Suma/Nap) sodium or placebo.
Figure 3 shows the within-patient consistency data. Pain-free response 2 h postdose was reported in at least two of the first three S/NS-treated attacks (with possible interspersed placebo treatment) in 55.0% of patients in Study 1 and 52.1% of patients in Study 2. Results were similar for 2hPF postdose in two of three attacks with no interspersed placebo treatment (data not shown).

Within-patient consistency data. Percentage of patients with pain-free response 2 h postdose and sustained pain-free response through 24 h postdose in at least one of the first three, two of the first three, and three of the first three attacks treated with sumatriptan/naproxen sodium.
Sustained pain-free response from 2 to 24 h postdose
Across attacks in both studies, sustained pain-free response from 2 to 24 h postdose (a co-primary end-point) was reported in more attacks after treatment with S/NS than with placebo (Study 1: S/NS 37%, placebo 17%; difference 20%; 95% CI 15, 27;
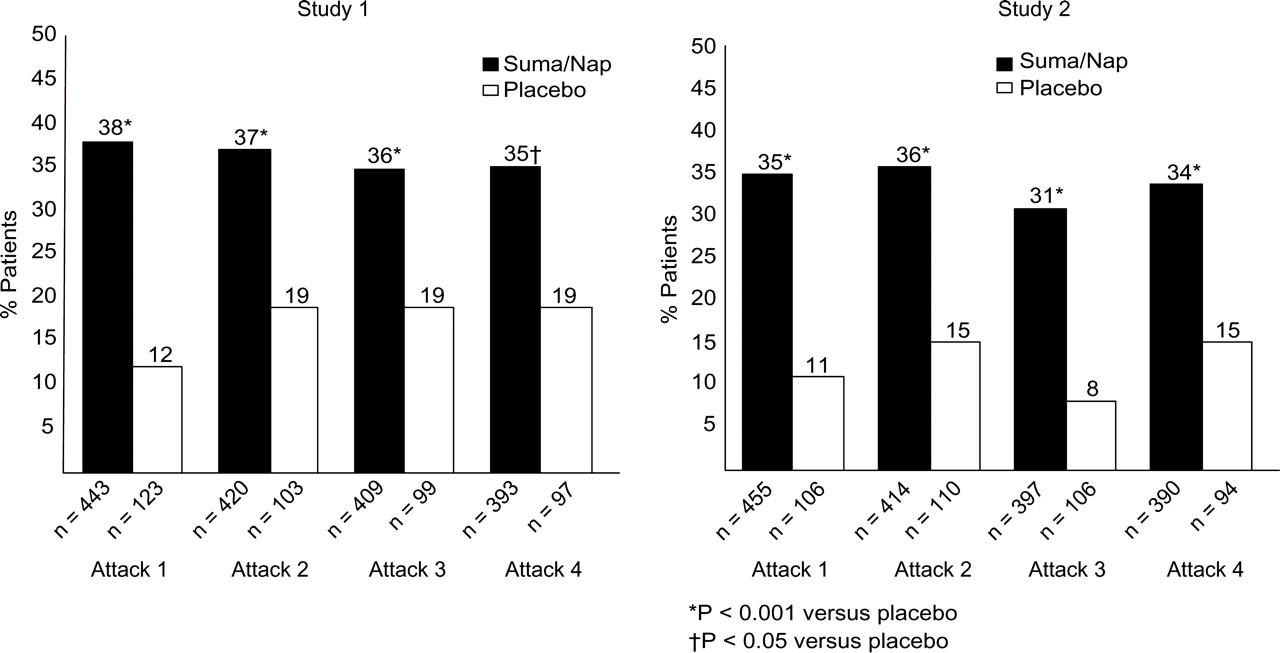
Twenty-four-hour sustained pain-free response after dosing with sumatriptan/naproxen sodium or placebo.
Sustained pain-free response 2–24 h postdose was reported in at least two of the first three S/NS-treated attacks (with possible interspersed placebo treatment) in 35.7% of patients in Study 1 and 32.6% of patients in Study 2 (Fig. 3). Results were similar for sustained pain-free response in attacks with no interspersed placebo treatment (data not shown).
Other efficacy measures
Across attacks in both studies, migraine-free response (i.e. no pain, nausea, vomiting, photophobia or phonophobia and no use of rescued medication) 2 and 4 h postdose was reported in more attacks after treatment with S/NS than with placebo (Table 2). Photophobia, phonophobia and nausea were reported in fewer attacks after treatment with S/NS than with placebo 2 h postdose (Table 2). Data for each attack considered separately were similar to the results summarized across attacks (Table 2). The use of rescue medication within 24 h of dosing with study medication was reported in fewer patients after treatment with S/NS than with placebo in both studies (
Secondary efficacy measures
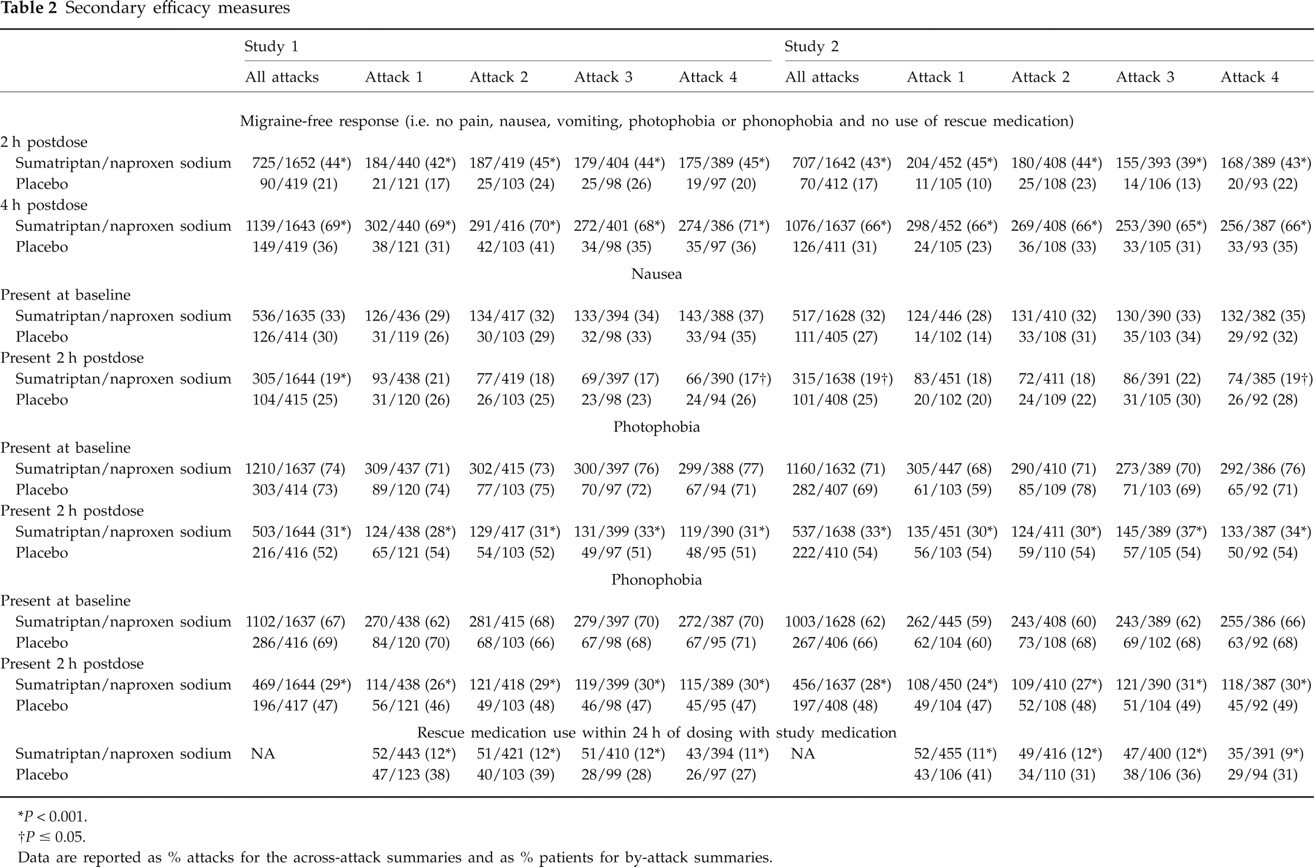
∗
†
Data are reported as % attacks for the across-attack summaries and as % patients for by-attack summaries.
Adverse events
In Study 1, the incidence of adverse events across attacks was 9% (153 of 1693 attacks) after S/NS treatment and 7% (28 of 424 attacks) after placebo treatment. In Study 2, the incidence of adverse events across attacks was 13% (219 of 1678 attacks) after S/NS treatment and 9% (36 of 422 attacks) after placebo treatment. Table 3 shows the incidence of individual adverse events reported in ≥ 2% of patients within 72 h of treatment with S/NS by attack.
Number (%) of patients with adverse events occurring within 72 h of treatment with sumatriptan/naproxen sodium in ≥ 2% of patients
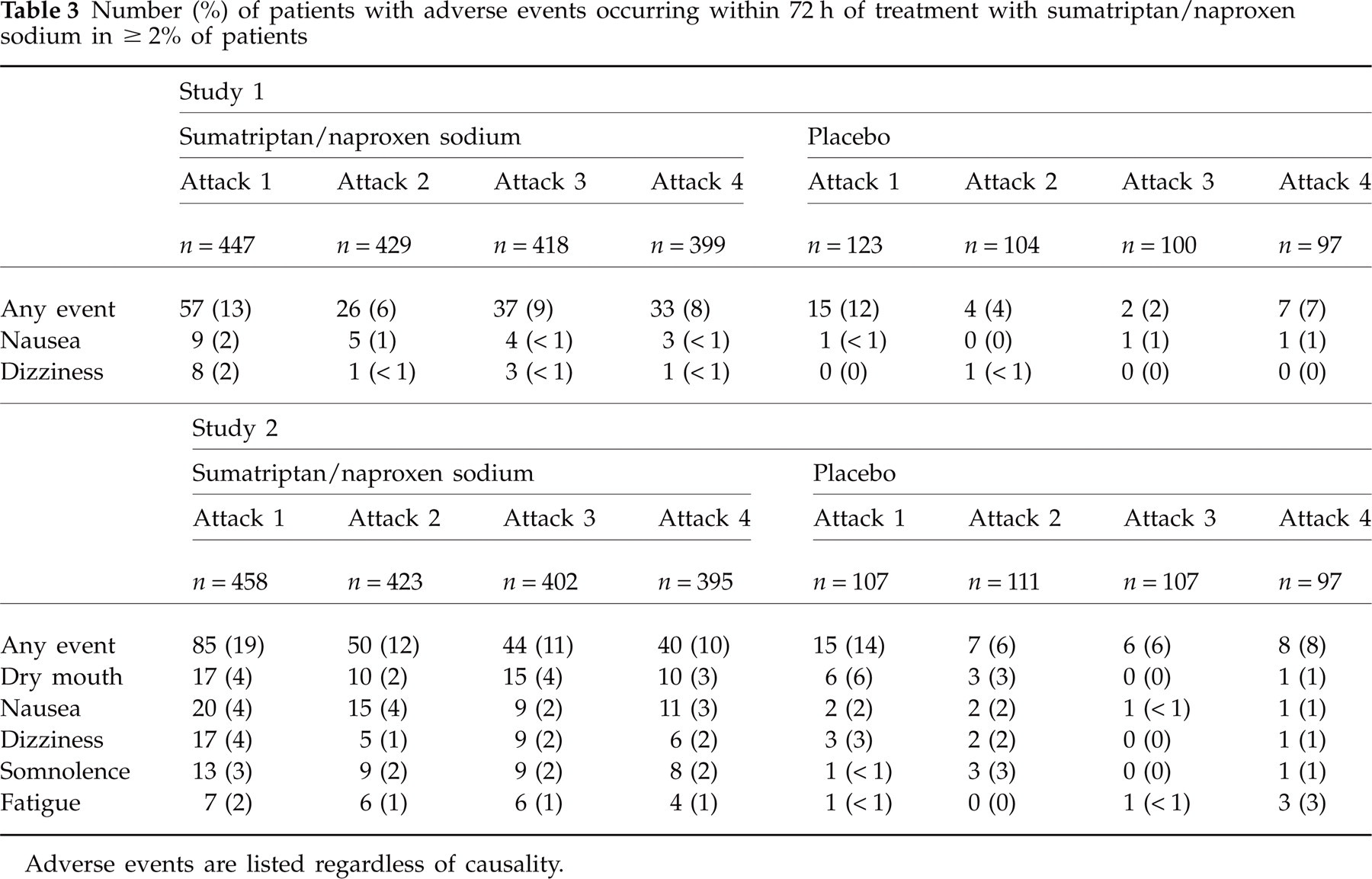
Adverse events are listed regardless of causality.
Seven treatment-emergent serious adverse events were reported across studies: ectopic pregnancy, ovarian cyst, severe viral gastroenteritis, bronchitis, breast cancer
In Study 1, the percentage of patients who withdrew because of adverse events occurring within 72 h of treatment was 1% with S/NS (
Discussion
Most migraineurs experience one or more migraine attack per month and use their acute migraine medication to treat many attacks. Therefore, consistent efficacy and tolerability over multiple migraine attacks are important attributes of acute therapy. The multiple-attack, random insertion of placebo design used herein has two major analytic benefits. First, by summarizing across attacks within person, more stable estimates of response to treatment are derived. Second, within-person consistency of response can be examined. The results of these two identical, randomized, double-blind, placebo-controlled studies demonstrate that S/NS used for a series of migraine attacks was effective and generally well tolerated across attacks. The therapeutic gain (i.e. the benefit of active treatment over that of placebo) was high across many outcome measures including both of the co-primary end-points. For 2hPF, the therapeutic gain was 28% in Study 1 and 30% in Study 2. For 24hSPF, therapeutic gain was 20% in Study 1 and 22% in Study 2. S/NS conferred high pain-free response during the 2-h postdose period and over 24 h. The data inspected for each attack reveal consistent response rates from attack to attack with no evidence of tolerance to the therapeutic benefits. These studies also demonstrate that S/NS relieves the associated symptoms of migraine, including nausea, photophobia and phonophobia.
Within-person consistency of response to S/NS was also demonstrated over multiple migraine attacks: in at least two of the first three attacks treated with active medication, more than half of patients were pain free 2 h postdose and approximately one-third had sustained 2–24-h pain-free response. The data suggest that S/NS may help to address patients' desire for a migraine therapy that provides consistent efficacy across multiple attacks. Comparable data on within-person consistency of response to other migraine therapies are not available from randomized trials using an early treatment design.
S/NS was generally well tolerated when used for approximately 3300 migraine attacks in 1135 patients in these two studies. The incidences of any adverse event and of specific adverse events were low and generally similar between S/NS and placebo. Furthermore, the incidence and pattern of adverse events did not change with use of S/NS over a series of attacks. The type and frequency of adverse events reported after treatment with S/NS in the current studies are consistent with those reported in previous studies of S/NS (17, 18) and studies of sumatriptan monotherapy or naproxen sodium monotherapy (25, 27, 29, 30).
These studies help to define further the therapeutic profile of S/NS, a combination treatment designed to target multiple mechanisms of migraine (17). When used to treat migraine with moderate or severe pain, S/NS has been demonstrated to be more effective than sumatriptan or naproxen sodium monotherapy in randomized, double-blind, single-attack studies (16) and to be generally well tolerated in a 12-month, open-label safety study (18). The current studies extend these results by demonstrating that the single-attack efficacy of S/NS is maintained with intermittent use over time and by corroborating the previous tolerability data.
The multiple-attack crossover design with random insertion of placebo has been used in only a few previous studies in migraine (6–9). The studies reported here, to the best of the authors' knowledge, are the first to use this design in the context of an early treatment paradigm. As treatment within 1 h and while pain was mild is widely recommended as an approach to treatment (19–25), this design has greater clinical relevance than previous studies in which moderate or severe pain was treated (6–9). The co-primary end-points of the current studies (2hPF postdose and sustained pain-free response 2–24 h postdose across attacks) provide a more stringent test of consistency of response than that of previous studies (6–9). Also, this study included the end-point of migraine-free response, which provides a more complete evaluation of an acute treatment of migraine than end-points assessing headache pain only. Furthermore, whereas both the previous and current studies assessed response to therapy during the early postdose period (i.e. 2 h postdose), the current studies also assessed pain-free response through 24 h after treatment in order to gauge consistency of sustained therapeutic response. The current studies also differ from previous placebo-controlled, crossover studies (6–9) in prospectively incorporating within-subject assessments of consistency of response. The previous studies, in contrast, focused primarily on between-subject assessments, although
These studies have demonstrated that use of the multimechanism migraine therapy S/NS in the early treatment paradigm may be an effective treatment option for achieving consistent relief from acute migraine attacks.
Footnotes
Competing interests
R. B. L. has consulted for or conducted research funded by Advanced Bionics, Allegan, Inc., Cierra, GlaxoSmithKline, Eli Lilly, Merck, Neuralieve, Novartis, Ortho-McNeil Neurologics, Pfizer, and Pro-ethics. D. W. D. is a consultant/advisor for Allergan Inc., Eli Lilly, GlaxoSmithKline, Merck, Neuralieve, Ortho-McNeil, Addex, Coherex, Pfizer, St Jude, and Solvay. He has received research support from Advanced Neurostimulation Systems and Medtronic. R. G. K. is a consultant for GlaxoSmithKline and Ortho-McNeil and has received honoraria from GlaxoSmithKline, Ortho-McNeil, Allergan, and Merck and Co. J. U. A. is a consultant/advisor for GlaxoSmithKline. He has received research support from AstraZeneca, Eisai, Ortho-McNeil, Pfizer, Merck, Vernalis, Pozen, and UCB Pharma. He is currently on speaker bureaus for GlaxoSmithKline, Merck, and Ortho-McNeil. S. E. L., A. C. N. and J. D. W. are employees of GlaxoSmithKline.
Acknowledgements
GlaxoSmithKline was the study sponsor and funded the research described in this manuscript. Pozen, Inc. was the IND sponsor of the research described. Author J. D. W. conducted the statistical analysis. The authors acknowledge Jane Saiers, PhD, for assistance with writing this manuscript. GlaxoSmithKline funded Dr Saiers' work. The authors acknowledge Mary S. Richardson, PharmD and Michael H. Ames, PhD, for input into the design of the clinical studies, and Susan A. McDonald, MA, for interpretation of the data and critical review of the manuscript.