
Abstract
There is an obvious need to improve clinical trial designs with respect to efficiency, duration and the number of patients recruited. Adaptive (flexible) designs may be valuable in this respect. We simulated the properties of a two-stage adaptive proof-of-concept and dose-finding trial design in adult migraine patients with moderate to severe headache, with or without aura. We also assessed the usefulness of a combined Bayesian and frequentist approach in the estimation of the probability of success of subsequent Phase III studies. Applying such an innovative approach would result in a reduction of the required sample size by 30 patients and no prolongation of the trial duration. The probability of success in Phase III is > 81%. An innovative adaptive design can facilitate testing of investigational migraine medications by reducing patient numbers and improving predictivity of success in Phase III.
Introduction
Migraine, a common disorder with a 1-year prevalence of 12%, is a leading cause of disability (1–3). Although medical management of migraine patients has improved significantly with the introduction of selective serotonin 5-HT1B/1D receptor agonists (triptans), there is still significant room for improvment. Many patients do not respond to triptans and response is often only partial (4). A number of patients becoming pain free 2 h after dosing have a relapse, i.e. re-occurrence of the migraine symptoms within 48 h, necessitating further treatment. As a result, a number of new and innovative pharmacotherapeutic approaches to the management of migraine are currently being persued.
The duration of drug development from discovery to registration has steadily increased over the years (5), resulting in a clear need for improvement in drug development programmes in terms of time and efficiency. One method proposed is the utilization of adaptive or flexible clinical trial designs. An adaptive design study is defined as a prospective study allowing for future planned design modifications during the performance of the study, dependent upon data accrued during the trial up to a defined interim time point (6, 7). Adaptations may include addition or removal of dose levels, re-estimation of sample size or randomization ratio, change of statistical methods, change of hypothesis, etc. Adaptive designs may also allow for a seamless transition from one phase of clinical development to the next, within a single trial (8–14).
The European Medicines Agency (EMEA) states that such an approach can speed up the process of drug development or can allocate resources more efficiently without lowering regulatory standards (15). Nevertheless, it must be noted that the application of adaptive design is connected to several methodological issues (16–23), of concern to clinical investigators, statisticians and regulatory authorities.
Although the concept of adaptive trial designs is several decades old, it has been implemented in only few migraine trials to date. This is surprising considering that in migraine clinical trials the clinical end-point (pain relief/pain free) is rapidly available after dosing. This is in contrast to other indications such as cardiac diseases or stroke, for which the read out in a clinical trial is available only with a certain time lag. Timely interim analysis while recruitment is still ongoing could therefore allow the flexibility of the adaptive-design approach to materialize.
In order to gain a better understanding of the potential of an adaptive clinical trial design in migraine, the combination of a proof-of-concept (PoC) and a dose-finding study for a putative new migraine compound in an adaptive design was compared with a tradional approach. Specifically, it was determined whether application of such an innovative design would result in reductions in sample size and duration. The primary objective of the adaptive trial would be to show efficacy of the new compound in a confirmatory sense (i.e. with an established P-value) to allow decision making to progress to Phase III. The practical constraints of such a programme were also considered.
Although the first application of Bayesian decision procedures in clinical research is almost 40 years old, there has been recent renewed interest in its application to early-phase clinical development (24). The benefits of application of a combined frequentist and Bayesian approach are evaluated. By combining results of the simulated findings of an interim analysis and the assumptions for the pivotal studies, an estimation of the probability of success in Phase III is possible. Based on this approach, the likelihood of an earlier trigger of Phase III is assessed.
Methods
Decision tree
Two conventional Phase II trials (a PoC and a dose-finding study) are combined through implementation of a two-stage adaptive design (9). The study consists of two stages separated by an interim phase. In stage 1, patients are randomized to either placebo or one of several active dose groups. Following the last patient, interim analysis follows. The data are analysed applying either an overall test or pairwise comparisons leading to a one-sided P-value (P 1). According to the decision tree (Fig. 1), the study stops at stage 1 for either futility (P 1 ≥ α0) or efficacy (P 1 ≤ α1). In all other cases, doses with no or minimal efficacy or with poor tolerability are pruned and only one active group and placebo proceed to stage 2 (the whole procedure can easily be adapted so that more than one active dose group is taken to stage 2). Patients’ recruitment continues up to the pre-planned sample size. Data from stage 2 are analysed using the same or a different statistical method, resulting in a one-sided P-value (P 2). The findings of both stages are combined through a combination function in order to control for the inflation of Type I error (α). Several publications summarize different combination techniques, but the modified Fisher's product test is preferred here for its properties and simplicity (25–30). From the comparison of the weighted product test P 1 w · P 2 with the critical value ca2 the intersection hypothesis H01 ∩ H02 is either rejected or accepted.

Decision tree in an adaptive two-stage procedure.
The two-stage adaptive design
Based on the above decision tree, several two-stage adaptive design procedures were considered for simulations, each differing only in respect of the decision rules applied for the selection of the ‘most promising’ dose (31–37). The option with the most prominant decrease in study duration and sample size was utilized as follows: patients were randomized to one of five active dose groups (standardized doses 0.10, 0.25, 0.50, 0.75 and 1.0) or placebo (dose 0). A Cochran–Armitage (C-A) trend test was applied at stage 1 (P 1) (38). Spending of α was equal at both stages (α1 = α2 = 0.02218). As long as dose proportionality was shown (P 1 ≤ 0.02218) the study was stopped for efficacy. Furthermore, the study was stopped for futility if P 1 ≥ 0.30 (α0). The C-A trend test defined the boundary α0. A P-value ≥ 0.30 corresponded to a random dose response pattern. In case that stage 1 was inconclusive (α1 < P 1 < α0), only the ‘most promising’ dose and placebo proceeded to stage 2. For the simulations the highest dose was assumed to have poor tolerability and hence was pruned, whereas the second highest was considered to be the ‘most promising’ dose (possible techniques involved in the selection of the ‘most promising’ dose(s), however, are not the subject of this review and are not discussed further). At stage 2, Fisher's exact test was applied to test superiority of the investigational drug vs. placebo (P 2). The value of weighted product test P 1 w · P 2 was compared with the critical value ca2 = 0.0033. As weight, w, the ratio of patients randomized at stage 1 vs. overall was taken. For all cases, the methodological properties were tested through simulations of 10 000 trials. The proportion of times that either the null hypothesis H01 or the intersection hypothesis H01 ∩ H02 was rejected was counted. The finding was an empirical estimate of the power of the study.
Estimation of sample size
For an initial estimate of the sample size required for selecting the ‘most promising’ dose after stage 1, the following three concepts were considered. According to Lang et al., a comparatively large stage 1 is necessary in order to obtain estimates that are sufficiently precise, and to take advantage of stage 1 as a learning stage (26). In contrast, Lachin et al. have recommended a range of 10–15 patients as being sufficient for stage 1 (39). Finally, Whitehead, applying a Bayesian approach, has shown that in the case of a pre-specified total number of treated patients, the probability of selecting the most promising dose group is maximized by taking a wide dose range with as few as five to six patients per dose group (40–42). As a compromise, the initial number of evaluable patients used in our simulations for stage 1 was between 10 and 25 per dose group. The sample size at stage 2 was determined through simulations by an attempt not to exceed the total number of patients required by a conventional design and the need to retain power at ≥ 0.80.
Assumptions for simulation
To initiate the simulation procedure, assumptions on the expected treatment difference, the dose–response relationship and the recruitment rate were made.
Assumptions on treatment difference
Trial methodology for acute treatments for migraine is well established. Guidance documents have been published by the International Headache Society (43) and the EMEA (44). The patient population and end-points used in the current simulations are consistent with the suggestions and requirements described in these documents. For example, efficacy was investigated in patients experiencing fully established moderate to severe migraine headache. The primary end-point of being pain free at 2 h was used.
Triptans are the gold standard for the treatment of migraine. Based on published results, 16–40% of treated patients are pain free at 2 h (4, 45). This compares with 3% and 15% of placebo-treated patients (46–56). For our study, a difference of 20–30% between at least one active dose group and placebo was assumed. The placebo effect was set at 10%.
Assumptions regarding the dose–response relationship
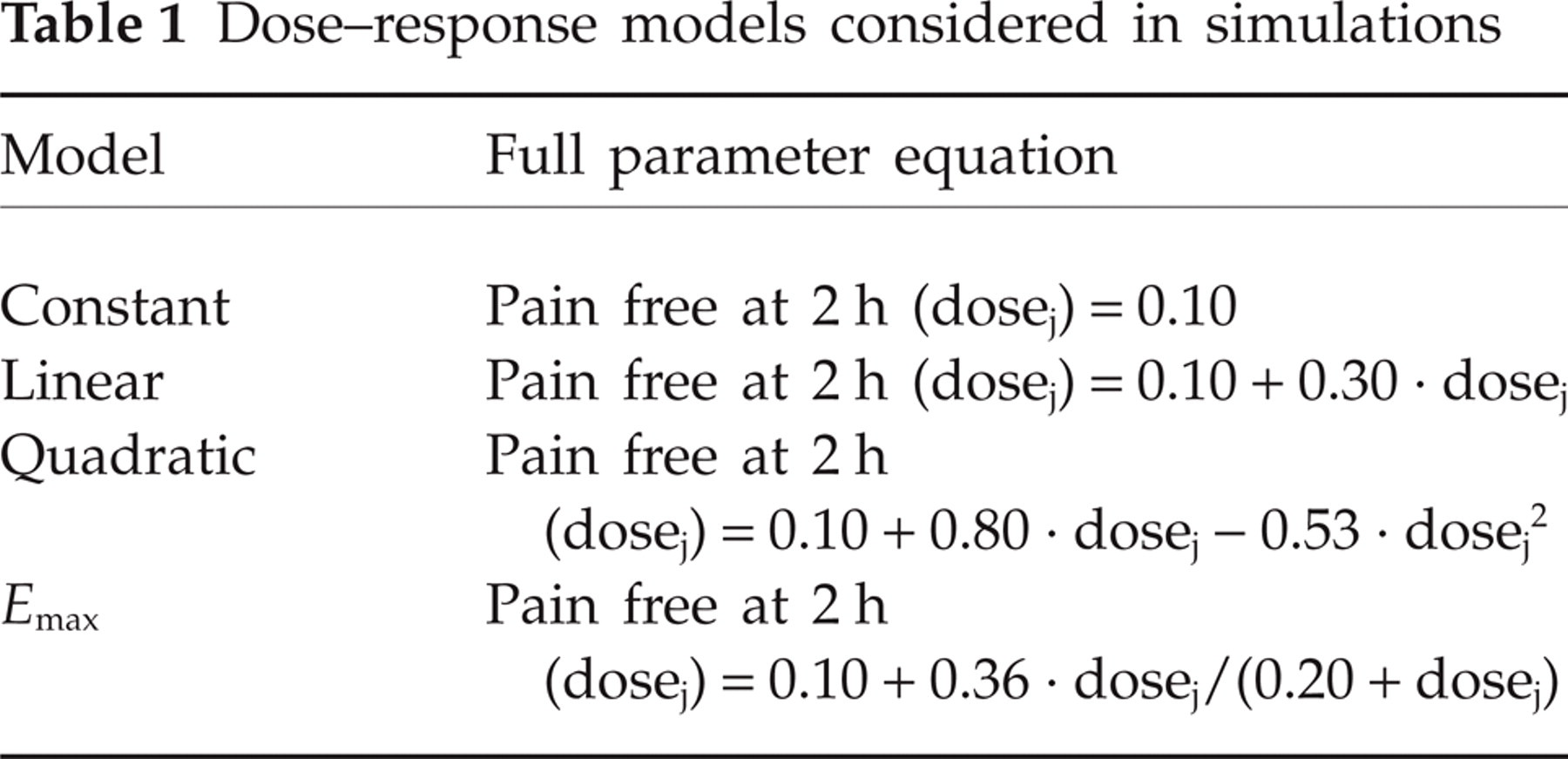
Several standard pharmacokinetic/pharmacodynamic (PK/PD) models are used to describe the dose–response relationship of pharmaceutically active compounds. In the present study three models were considered: linear, quadratic and E max. A maximum effect of 40% was used for all three models. For the quadratic model an optimal dose of 0.75 was used. For the E max model half of the maximum change in effect (ED50) was set at the dose level 0.20. As a control model, a constant effect was also considered, assuming no treatment gain of the active drug over placebo (Table 1).
Dose–response models considered in simulations
Assumptions for recruitment rate
We assumed that 25 sites would take part in the study. The recruitment rate was estimated to be two patients per month and site. The period between the last patient in for stage 1 and the end of the interim analysis was assumed to be 1 month. During this time, recruitment would be controlled to four patients per arm.
Results
The features of the outlined two-stage adaptive design with comparison with a conventional development approach in terms of patient numbers and time are provided. The benefits of applying a combined frequentist and Bayesian approach for the development of a new migraine medication are also provided.
Innovative design
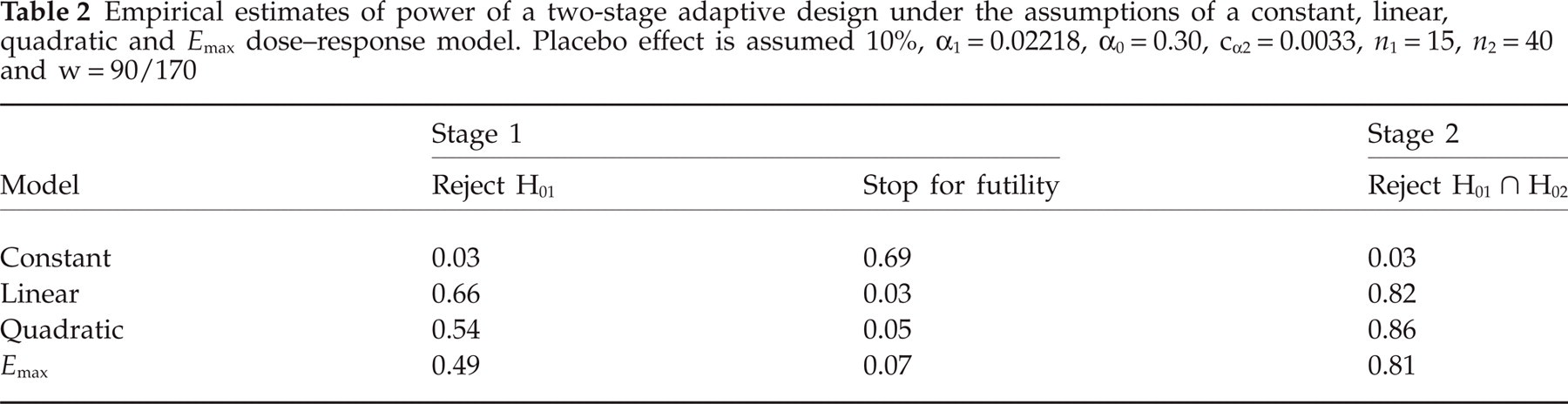
Fifteen evaluable patients per dose group were randomized at stage 1. A significant dose–response trend (C-A trend test, P 1) could be shown in ≥ 49% of the cases, whereas the study stopped for futility in ≤ 7%. In those cases where the outcome was indecisive, placebo as well as dose group 0.75 proceeded to stage 2. Based on the conducted simulations, 40 (40) evaluable patients per arm were planned for the second part of the two-stage adaptive design. Superioriry vs. placebo was tested using Fisher's exact test (P 2). The product P 1 0.53 · P 2 was compared with the critical value cα2 = 0.0033. The estimated power ranged between 81% and 86%, depending on the applied PK/PD model (Table 2).
Empirical estimates of power of a two-stage adaptive design under the assumptions of a constant, linear, quadratic and E max dose–response model. Placebo effect is assumed 10%, α1 = 0.02218, α0 = 0.30, cα2 = 0.0033, n 1 = 15, n 2 = 40 and w = 90/170
In total, 186 evaluable patients would have to be randomized accounting for a continuation of the recruitment during the interim phase. Fifteen patients per arm were randomized at stage 1 (Nstage 1 = 15 × 6 = 90). Sixteen cases were randomized to four pruned doses plus eight cases in placebo and the ‘most promising’ dose group during the interim analysis phase (Ninterim = 4 × 6 = 24). Seventy-two evaluable patients per group were planned to be randomized after the interim phase and during stage 2 (Nstage 2 = (40 − 4) × 2 = 72).
In the hypothetical scenario that the two higher doses would fail due to tolerability issues, while the next lower dose (0.5) and placebo would continue to stage 2, the power of the study would still be close to the desired level (≥ 0.73). Concerning the duration of the trial, the complete study (stage 1 and 2) could be conducted within 18 weeks. Should the study stop early (for efficacy or futility; only stage 1), the duration would be 12 weeks.
Conventional design
The minimum number of evaluable patients required to show a statistically significant dose–response trend under the assumption of the three PK/PD models was 32 patients (one-sided C-A trend test; α= 0.025; 1 − β= 0.80). Considering that six dose groups (five active and placebo) would be included, 192 evaluable patients would have to be randomized.
Furthermore, at least 36–69 evaluable patients per arm are needed to detect the difference of 30–20% between the ‘most promising’ dose and placebo as statistically significant with power 80% and α= 0.025 (one-sided Fisher's exact test).
Consequently, 216–414 cases would have to be randomized in a single Phase II study in order to address both objectives, PoC and dose response. Conduct of a conventional design study can hence be assumed to require 18–33 weeks, depending on the sample size.
Combination of a frequentist and a Bayesian approach
We applied the approach described by Stallard et al., i.e. preparation for Phase III starts when the Pr (probability for success in Phase III) is greater than or equal to a pre-specified critical value p u(57). Thall et al. have presented a similar approach (58). The assumptions on the probability distribution (often called prior) β(α,β) with α + β= 2 that expresses our uncertainty about the percentage of patients pain free at 2 h before the interim information is taken into account was based on the publication of Thall and Simon (59). More precisely, the response probability of the ‘most promising’ dose and placebo was taken to have a prior β(0.6,1.4) and β(0.2,1.8), respectively. These priors correspondede to weak suggestion of pain free at 2 h in 30% and 10% of the cases. The critical value p u was justified through simulations of 10 000 trials (Table 3).
Probability of success in Phase III after the interim analysis for a range of p u critical values, for all PK/PD models

The choice of a small p u value increased the probability of success of the Phase III programme. In contrast, a larger value for p u contained the risk of abandoning the development of an efficacious substance. In our case, the critical value p u was taken equal to 0.50. The findings in Table 3 denote a high probability of initiating Phase III based on the outcome of the interim analysis.
Discussion
There is an obvious need for efficient clinical trial designs and drug development strategies. Such innovative designs may reduce the time until a new migraine drug becomes available to patients. In addition, such an approach may reduce the number of patients included in a clinical study and reduce exposure to ineffective compounds or ineffective doses. In the area of migraine, few studies with innovative adaptive designs have been reported (60–64). Apart from the lack of experience with such flexible designs in migraine clinical trials, other issues such as maintainance of trial integrity and increased planning complexity (blinding, drug supply, interim analysis, rapid turnaround, etc.) must be considered. A clear understanding of the potential and risks of adaptive study design for drug development in migraine is mandatory.
The focus of the current study was to investigate two questions: to what extent could a two-stage adaptive design allow reduction in the number of patients exposed, and would such an approach have the potential to reduce the time required for the early phase of clinical development? The simulated findings were compared with the outcome of a classical approach. With a conventional design, at least 216 evaluable patients have to be randomized in order to show superiority of one active dose group over placebo with statistical significance with an expected study duration of 18 weeks. Alternatively, by utilizing a two-stage adaptive design, 114–186 patients are necessary and the Phase II study can be assumed to last 12–18 weeks. This approach has important ethical implications, since fewer patients have to take part in the experiment, fewer cases are exposed to placebo, and fewer patients are exposed to a non-active compound and non-active doses. There is also the potential for important time savings.
The implementation of an interim analysis as simulated here does not only have the advantage of terminating the study early. By using the data accrued during stage 1, preparation for the Phase III programme can be triggered earlier. To make use of this benefit, a combined frequentist and Bayesian approach was used. By combining the stage 1 simulated findings with the assumptions made for the pivotal studies, the probability of success in Phase III was > 81%.
However, when simulating and planning for such a trial, several practical issues have to be taken into consideration.
One important issue is related to the data of patients randomized to the pruned groups (16 patients) during the interim phase. These data would not be included in any analysis. This is problematic in two ways. First, from a methodological point of view it is problematic to omit any generated data and hence is in clear contradiction to authorities’ guidance, requesting inclusion of all treated patients. Second, from an ethical point of view it is not acceptable to treat patients in a clinical study without making use of the data generated. This issue, however, can in our view be solved by integrating these patients in the analysis of stage 1.
Another issue is related to the communication of the findings of the combined frequentist and Bayesian concept. This approach carries the risk of damaging the integrity of the study, since starting any preparations before the completion of the study would automatically signal a positive outcome. Therefore, in our opinion such an approach has to be applied after the completion of stage 2.
The inclusion of more than one interim analysis was also considered, but was found to be not practical since it whould automatically result in a prolongation of the Phase II programme.
Another issue is related to the involvement of an independent data monitoring committee (DMC) with full authorization in the decision-making process. Our position is that participation of an external DMC increases the reliability and credibility of trial results. Any pre-specified algorithm defined by the sponsor should rather serve as guidance rather than as a strict rule. This recommendation is based on the rationale that early data often involve unexpected information that cannot be easily modelled in any simulation. Therefore, experts’ opinion on eliminating dose groups and on trial continuation must be considered as especially valuable.
The consequences of early termination due to efficacy were also assessed. Early termination may be challenged, given that the information gained during stage 1 is generally insufficient regarding secondary efficacy end-points. However, in the case of migraine, where the primary end-point is very well defined, in-depth investigation of secondary efficacy outcomes can be left to subsequent studies. At this early stage the primary interest should be to show overall efficacy and to select the ‘most promising’ dose(s) for subsequent studies.
Another risk of stopping the study for efficacy at stage 1 is the resultant reduction in the safety data prior to entry to Phase III. Data from a study as large as that including stage 2 may be required to initiate Phase III safely. In addition, safety from higher doses than could be achieved in the traditional clinical study approach would also provide safety bracketing at a higher than therapeutic dose. However, the present study has shown that ≥ 95 patients (15 evaluable patients ×5 active dose groups during stage 1 plus 20 patients during interim phase) are foreseen to be randomized and treated during the first stage of the study with the active compound. We believe that this number is sufficient to describe adequately the tolerability profile of an investigational Phase II drug.
The greatest risks involve the large number of assumptions made in laying out of the comparison of adaptive and traditional study designs. More precisely, in a confirmatory PoC study assumptions are generally made regarding the expected treatment difference (Δ) based on literature review, whereas in a dose-finding study assumptions are made with respect to the possible pattern of the dose–response curve based on pharmacological PK/PD modelling. In the innovative adaptive design approach, additional assumptions and design features are included that could have an impact on outcome. For example, different scenarios and assumptions were tested regarding the combination test used, the splitting of Type I error, the number of treated patients in the two stages, the number of interim analyses, the applied statistical tests, and the selection of the ‘most promising’ dose. The proposed design is a result of numerous simulations and combination of several different assumptions, each of which carries some risk of uncertainty.
In conclusion, the described two-stage adaptive design, under the assumed conditions, may be a promising tool in early clinical development of new migraine drugs to reduce the number of patients exposed to experimental therapy and reduce the time to Phase III. Benefits and risks of adaptive design will vary greatly based on the individual mechanism and disease involved and underlying assumptions. As a result, the decision to use adaptive design and the specific design to be used can be made only on a case-by-case basis after discussions between all involved parties, including clinical pharmacology, modelling and simulation specialists, statisticians, investigators and regulators.
Footnotes
Acknowledgements
The authors wish to thank Dr H. Peil, Dr A. Gupta and the referees for their helpful comments and suggestions.