
Abstract
Introduction: Lasmiditan (COL-144; LY573144) is a novel, highly selective and potent agonist at 5-HT1F receptors that lacks vasoconstrictor activity. Preclinical and early clinical experiments predict acute antimigraine efficacy of COL-144 that is mediated through a non-vascular, primarily neural, mechanism.
Subjects and methods: In a randomised, multicentre, placebo-controlled, double-blind, group-sequential, adaptive treatment-assignment, proof-of-concept and dose-finding study, we treated 130 subjects in-hospital during a migraine attack. Subjects were allocated to an intravenous dose level of lasmiditan or placebo in small cohorts. The starting dose was 2.5 mg. Subsequent doses were adjusted, up or down, according to the safety and efficacy seen in the preceding cohort. The primary outcome measure was headache response defined as improvement from moderate or severe headache at baseline to mild or no headache at 2 h post-dose. The study was designed to explore the overall dose response relationship but was not powered to differentiate individual doses from placebo, nor to detect effect differences for other migraine symptoms.
Results: Forty-two subjects received placebo and 88 received lasmiditan in doses of 2.5–45 mg. Subjects were observed in the clinic for 4 h after treatment and used a diary card to record symptoms and adverse events for up to 24 h. The study was terminated when the 20 mg dose met predefined efficacy stopping rules. Of subjects treated in the 10, 20, 30 and 45 mg lasmiditan dose groups, 54–75% showed a 2 h headache response, compared to 45% in the placebo group (P = 0.0126 for the linear association between response rates and dose levels). Patient global impression at 2 h and lack of need for rescue medication also showed statistically significant linear correlations with dose. Lasmiditan was generally well tolerated. Adverse events were reported by 65% of subjects on lasmiditan and by 43% on placebo and were generally mild. Dizziness, paresthesia and sensations of heaviness (usually limb) were more common on lasmiditan.
Conclusions: At intravenous doses of 20 mg and higher, lasmiditan proved effective in the acute treatment of migraine. Further studies to assess the optimal oral dose and full efficacy and tolerability profile are under way. The non-vascular, neural mechanism of action of lasmiditan may offer an alternative means to treat migraine especially in patients who have contra-indications for agents with vasoconstrictor activity. The clinicaltrials.gov identifier for this study is NCT00384774.
Introduction
Migraine is a common and highly disabling brain disorder, affecting over 10% of adults globally (1). The disease is typically characterised by attacks of 1–3 days of severe headache, associated with nausea, vomiting, photo- and phonophobia (migraine without aura) and, in one-third of patients, neurological aura symptoms (migraine with aura) (2).
The pathogenesis of migraine is incompletely understood. Traditionally, vasodilatation was considered pivotal in causing the headache in migraine (3). Triptans, selective 5-HT1B/1D receptor agonists with established antimigraine efficacy (4), were developed based on the assumption that 5-HT1B receptor-mediated cranial vasoconstriction is a prerequisite for antimigraine efficacy (5). As a consequence, triptans also carry the risk of causing coronary vasoconstriction (6) and are contra-indicated in patients with cardio- and cerebrovascular disease. In addition, many patients using triptans report chest symptoms, which may mimic angina pectoris, causing anxiety and diagnostic confusion (7,8). Thus, novel antimigraine treatments that are devoid of vasoconstrictor activity are warranted.
In recent decades, it has become evident that cranial vasodilation, if it happens at all during a migraine attack (9), may only be a secondary phenomenon due to activation of the trigeminovascular system (2). Vasoconstriction may thus not be necessary to treat migraine headaches. Rather, neural inhibition of trigeminal pathways would provide an attractive alternative non-vascular antimigraine mechanism. Indeed, LY334370, a neurally active selective 5-HT1F receptor agonist with no vasoconstrictor activity at clinically relevant concentrations, proved effective in the acute treatment of migraine in an early clinical proof-of-concept study (10). Unfortunately, the clinical development of LY334370 had to be stopped because of compound-specific safety concerns on long-term exposure in animals.
Lasmiditan (COL-144, formerly known as LY573144; Eli Lilly & Company) is a new selective and highly potent 5-HT1F receptor agonist, with a Ki at human 5-HT1F receptors of 2.21 nM and an affinity which is more than 470-fold higher for 5-HT1F receptors than for other 5-HT1 receptor subtypes (11). Lasmiditan, and other selective 5-HT1F agonists, are active in neurally mediated preclinical models of migraine, without causing vasoconstriction. These experiments demonstrate potent inhibition of c-Fos induction in the trigeminal nucleus caudalis and inhibition of dural plasma protein extravasation following electrical stimulation of the trigeminal ganglion. At concentrations up to 0.1 mM, lasmiditan did not constrict rabbit saphenous vein, a surrogate assay for human coronary vasoconstrictor liability (11).
Based on its pharmacological profile, and the promising early clinical data obtained with LY334370 (a prototype 5-HT1F receptor agonist), we believe that lasmiditan might provide acute relief of migraine via a novel neural mechanism of action without the potential drawbacks of associated vasoconstrictor activity. Here, we set out to test the efficacy of lasmiditan in the acute treatment of migraine and to obtain early information on the effective dose range. In order to maximise the clinical information, while keeping the number of study subjects as low as possible, we applied a group-sequential, adaptive-treatment design. This elegant and relatively new design allows for a quick, though reliable, screening for efficacy and broad tolerability at consecutive dose levels, and may significantly speed up the time to reach an effective dose level, while minimising the number of subjects being exposed to study drug or placebo.
Subjects and methods
Methods
The present study was a multinational, multicentre clinical trial conducted at 11 sites in Germany, four in Finland, and three in The Netherlands, between August 2006 and July 2007. The study was designed by the members of the Steering Committee together with the sponsor (CoLucid Pharmaceuticals Inc.). All authors had unrestricted access to the data. The study was conducted in accordance with the Declaration of Helsinki and internationally accepted standards of Good Clinical Practice. Prior to initiation, it was approved by the relevant regulatory authorities and independent ethics committees. All subjects gave written informed consent. The clinicaltrials.gov identifier is NCT00384774.
Study design
The study used a prospective, randomised, double-blind, placebo-controlled design with group-sequential adaptive-treatment assignment (12,13). Subjects were allocated to a dose level of lasmiditan in small cohorts, with the first 20 cohorts consisting of six subjects (4 received lasmiditan and 2 placebo) and subsequent cohorts of five subjects (4 lasmiditan and 1 placebo). The first cohort was allocated to the 2.5 mg dose level. The dose used in subsequent cohorts depended on the headache response (moderate or severe headache reduced to mild or none at 2 h) of the previous cohort: if two or less of the four active-treated subjects had responded, the dose was increased, and if three or more of the four active-treated subjects had responded, the dose was reduced. The dose adjustment rules were chosen to identify doses of lasmiditan with efficacy similar to, or better than, an oral triptan. This dose escalation or reduction sequence would be modified if two or more active-treated subjects in any cohort experienced a severe non-serious adverse event, in which case the dose would be reduced for the next cohort irrespective of the response rate. The occurrence of a drug-related serious adverse event would lead to automatic suspension of the randomisation pending a safety review. The lowest permissible dose of lasmiditan was 1 mg and the highest 60 mg.
The up-and-down dose adjustment process was terminated with the selection of an effective dose when the following criteria had been met: at least five blocks of subjects had been treated at this dose, and for at least four blocks the decision rule called for a dose decrease. Alternatively, the dose selection process could have been terminated, without the selection of an effective dose, if five consecutive blocks of subjects had been treated at the top dose with the escalation rules calling for a dose increase each time.
Subject screening and selection
Subjects were recruited either from the patient population at each site or by local advertising. Written informed consent was obtained and then subjects were screened for eligibility at an out-patient visit outside a migraine attack. Eligible subjects were invited to return to the clinic for treatment with study medication of a new, moderate or severe migraine attack within 4 h of onset. On return to the clinic, eligibility for the study was reconfirmed and, provided that the investigator considered the on-going attack to be a migraine and no previous treatment had been taken for that attack, the subject was randomised. Subjects were eligible for the study if they were between 18–65 years of age and had at least a 1-year history of migraine with or without aura fulfilling the International Headache Society (IHS) diagnostic criteria 1.1 and 1.2.1 (2004), with a migraine onset before the age of 50 years (14). Subjects had to be experiencing 1–8 migraine attacks a month and not be using migraine prophylactic medication. Subjects were in good general health and had no evidence of vascular disease or hypertension. Subjects with previous intolerance of triptans were excluded but triptan non-responders could be included. Pregnant or breast-feeding women were excluded, as were women of child-bearing potential who were not using a highly reliable form of contraception.
Study procedures
On return of the subject to the clinic, instructions for dilution of study drug were obtained from an online randomisation system by a pharmacist or other study personnel, independent of the investigator, and the study drug for infusion was prepared. Both investigator and pharmacist were blinded with regard to active or placebo and only the pharmacist knew the dilution. Active and placebo were identical in appearance and packaging. All subjects received a 60-ml intravenous infusion over 20 min. Efficacy and safety data before and after administration of study drug were entered immediately into an electronic data capture system, so allowing the headache response to be used to drive dose-allocation for subsequent cohorts.
After baseline assessments were completed, lasmiditan or placebo was infused intravenously over 20 min and the subject was monitored for safety and efficacy by means of frequent ECGs, vital signs and adverse event recording and standardised timed questions about migraine symptoms for at least 4 h. Haematology and clinical chemistry assessments were conducted at screening, during the acute attack and at follow-up. Data, including those collected by questioning the subject, were entered concurrently into an online electronic data capture system by the study staff. Subjects were discharged from the clinic after 4 h and continued to record migraine symptoms and adverse events until 24 h using a diary card. Rescue medication (not triptans) was permitted from 2 h.
Symptom evaluation
A number of different symptoms were evaluated. The severity of headache was measured on a 4-point scale with 0 = no pain, 1 = mild pain, 2 = moderate pain, 3 = severe pain. Associated symptoms (nausea, vomiting, photophobia, phonophobia) were recorded as present or absent. Disability was documented on a 4-point scale with 0 = no disability, 1 = mild disability, 2 = moderate disability, 3 = severe disability. Data for the patient global impression were collected on a 7-point scale with 1 = very much better, 2 = much better, 3 = a little better, 4 = no change, 5 = a little worse, 6 = much worse, 7 = very much worse.
The primary efficacy measure was headache response, defined as a reduction in headache severity from moderate or severe at baseline to mild or no headache at 2 h after initiation of infusion of study drug (15). The secondary efficacy measures were: (i) rates of headache response at 10 min, 20 min, 40 min, 60 min, 90 min, 180 min, and 240 min after initiation of study drug infusion; (ii) rates of headache-free (reduction from moderate or severe headache at baseline to no headache pain) at 10 min, 20 min, 40 min, 60 min, 90 min, 120 min, 180 min, and 240 min after initiation of study drug; (iii) rates of sustained response, defined as a moderate or severe headache at baseline which became mild or no headache at 2 h after initiation of study drug and which did not recur (become moderate or severe) within 24 h of initiation of study drug; (iv) rates of sustained pain-free, defined as a moderate or severe headache at baseline which became no headache at 2 h after initiation of study drug and which did not recur (become mild, moderate or severe) within 24 h of initiation of study drug; (v) presence of nausea, vomiting, photophobia and phonophobia, and degree of clinical disability throughout the study course; and (vi) proportion of subjects using rescue medication between 2–24 h after initiation of study drug, and patient global impression 2 h after initiation of study drug.
Statistical methods
The target sample size of at most 160 subjects, with at least 20 subjects treated with an effective dose level and at least 10 subjects treated with placebo, was selected to provide appropriate preliminary data on which to choose a dose range for further evaluation. The statistical properties of the hypothesis tests to compare one or more dose levels to placebo when doses are allocated using the group sequential adaptive treatment assignment design were not known. Formal statistical tests were, therefore, not used to declare the study to be ‘positive’ or ‘negative’ and the study was not powered for statistical significance. Furthermore, the sample size was not powered for statistical considerations.
At the conclusion of the study, the headache response rates were summarised by dose level. The Mantel–Haenszel test was used to test for a dose-response relationship. Since the study terminated due to selection of an effective dose, Fisher’s exact test was used to compare the headache response rates for the selected dose versus placebo. In all analyses, the results for each dose level (including placebo) were combined across all blocks where that dose was used.
All subjects who received any study medication were included in the analysis population. The subjects were analysed according to the treatment and dose level they actually received which was in every case that to which they were randomised. Missing values were not replaced.
Results
Subject population
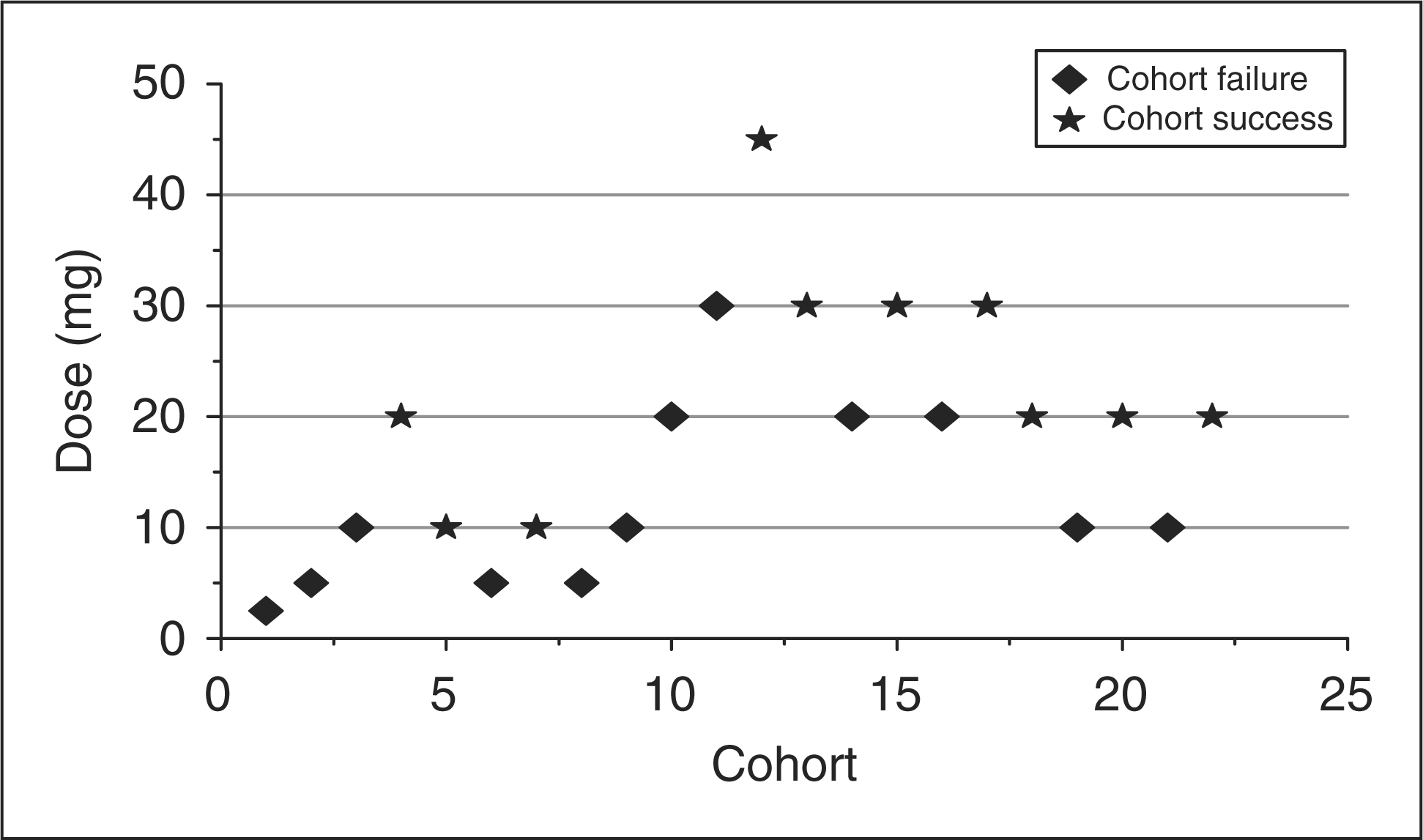
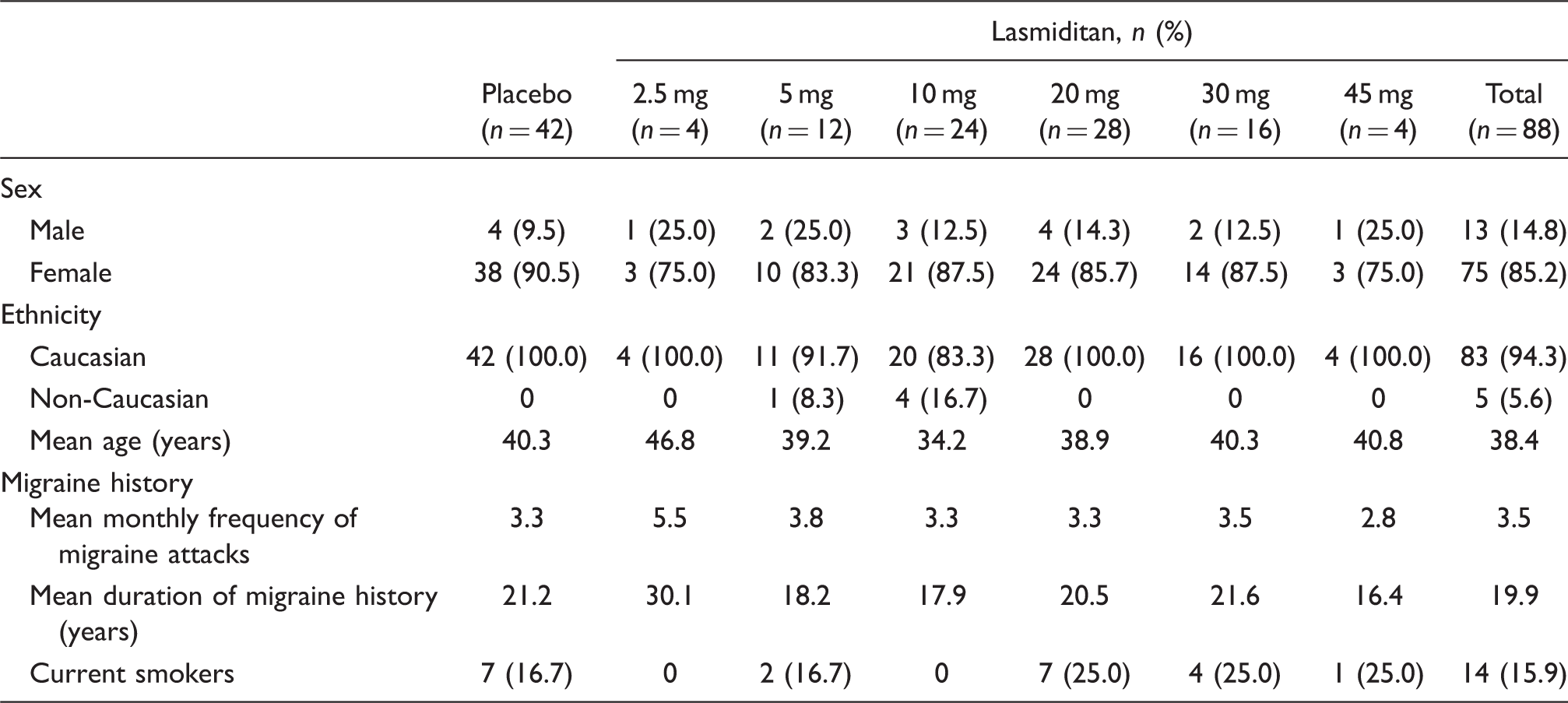
In total, 372 subjects were screened at 18 centres in Finland, Germany and The Netherlands and 130 returned for treatment in the clinic. These 130 subjects made up the analysis population. The treatment groups were generally well matched for demographic and baseline characteristics for the analysis population (Table 1). The majority of subjects were female in both treatment groups: ratio F:M for lasmiditan 6:1, and for placebo 10:1. The majority of subjects were Caucasian in both treatment groups (lasmiditan 94.3%, placebo 100.0%). Subjects were between 19–63 years old, with a mean age of 38.4 years in the lasmiditan group and 40.3 years in the placebo group. Subject disposition is shown in Figure 1. The sequence of subject allocation to treatment groups is shown in Figure 2.
Subject disposition. Dose escalation sequence. Proportion of subjects with a headache response at 2 h. Bars represent percentage of subjects at each dose with headache response at 2 h. Numbers within bars represent the number of subjects that responded at 2 h (numerator) and the number of subjects who received that dose (denominator). Subject demographics and background characteristics: analysis population

Efficacy
The dose escalation was terminated after 130 subjects, when the predefined stopping rules identified 20 mg as an effective dose based on the results for the primary endpoint. A higher proportion of subjects showed a 2 h headache response in the 10 mg, 20 mg, 30 mg, and 45 mg lasmiditan dose groups (54.2–75%) compared to placebo (45.2%; Fig. 2). The linear association between response rate and dose level was statistically significant (P = 0.0126; Mantel–Haenszel test for trend). Due to insufficient power for comparing individual dose levels, no individual lasmiditan dose was statistically significantly different from placebo at the 2 h time point (Fisher’s exact test).
A similar trend for increasing efficacy with increasing dose was observed (though not statistically tested) for headache freedom at 2 h post dose (Fig. 3). In line with these findings, the proportion of subjects using rescue medication showed an inverse trend with dose.
Proportion of subjects with headache response (moderate or severe predose becoming mild or none) 10–240 min post dose

P-values = 0.048 (20 mg/180 min), 0.048 (20 mg/240 min), 0.009 (30 mg/40 min), 0.007 (30 mg/60 min), 0.036 (30 mg/180 min), 0.017 (30 mg/240 min), 0.014 (45 mg/10 min), Fisher’s exact test, dose group versus placebo.
Secondary efficacy parameters

Tolerability and safety
Proportion of subjects with adverse events by preferred term (frequency ≥ 5% in either placebo or total lasmiditan group)

Discussion
We tested the acute antimigraine efficacy of lasmiditan, a novel, highly selective and potent agonist at 5-HT1F receptors. Its effect is most likely mediated through a primarily neural and non-vascular mechanism. We used a relatively novel up-and-down dose-adaptive study design to minimise subject exposure to study drug or placebo while still rapidly and reliably screening for efficacy and tolerability across a wide dose range. We found clear dose-related efficacy of lasmiditan in the acute treatment of a migraine attack. The onset of headache relief was evident at 20–40 min after the start of a 20 min intravenous infusion. As lasmiditan is devoid of vasoconstrictor activity at clinically relevant doses, the results of this study confirm that vasoconstriction may not be a prerequisite for antimigraine efficacy as has been suggested earlier (10,16). This clearly opens the possibility of new treatment options for patients who cannot tolerate, or have contra-indications for, triptans.
Lasmiditan was well tolerated. There were no clinically significant abnormalities of any safety parameters, i.e. heart rate, blood pressure, 12-lead ECG, haematology, biochemistry and urine analysis, following administration of lasmiditan. No subject terminated treatment because of side effects. There were also no subject-reported triptan-like chest symptoms or chest discomfort.
The 20 mg and higher doses of lasmiditan were identified as doses of interest for further evaluation. Pharmacokinetic/pharmacodynamic modelling using pharmacokinetic data from this study will facilitate the selection of an active dose range for evaluation when given by non-parenteral routes of administration.
This study had a high placebo response rate, which is most likely due to the conditions under which the trial was conducted. Attendance at the clinic for treatment may have heightened subject expectations and trials involving parenteral administration of acute antimigraine therapies have historically often demonstrated higher placebo rates than those in which the drug was given orally (17).
We used an adaptive design to identify the lowest effective dose. This can be achieved with minimal subject exposure to ineffective low doses compared to a parallel group design, where the distribution of subjects to dose groups is predefined. Furthermore, choice of a low starting dose and gradual escalation with on-going safety monitoring ensured that risk to the subjects was minimised. The adaptive design does, however, limit the exploration of the top end of the dose response curve since the dose is adjusted downwards as soon as the target efficacy rate – in this case a headache response rate of 75% – is reached.
This trial did not use an active comparator. Comparison with an oral triptan, analgesic or anti-inflammatory drug would have necessitated a double-dummy technique to maintain blinding and the small and variable group size inherent in the adaptive design would not have given sufficient statistical power to make a valid comparison between individual lasmiditan dose levels and the comparator drug. Instead, the adaptive design was chosen to explore a wide range of doses and guide dose selection for future studies of oral lasmiditan by means of pharmacokinetic/pharmacodynamic modelling.
Conclusions
Selective 5-HT1F receptor agonists may offer an alternative means to treat acute migraine attacks without the perceived cardiovascular risk associated with triptans. Furthermore, the neural mechanism of action may offer efficacy advantages that will be explored further in future studies.
The European COL-144 (Lasmiditan) Investigators Group
Germany: H-C Diener (Essen), A Beckmann-Reinholdt (Königstein), S Evers (Münster), A May (Hamburg), U Reuter (Berlin), M Ribbat (Itzehoe), C Riemasch-Becker (Wiesbaden), S Schuh-Hofer (Tübingen), K Shakeri-Nejad (Berlin), H Staudenmayer (Göppingen), A Straube (München). Finland: M Färkkilä (Helsinki), M Illmavirta (Jyväskylä), E Säkö (Turku), M-L Sumelahti (Tampere). The Netherlands: M Ferrari (Leiden), K Burggraaf and R Klein (Leiden), W Mulleners (Nijmegen), R De Rooij (Utrecht).
Disclosures
MDF has, in the past 3 years, received grants and consultancy or industry support from Almirall, Coherex, CoLucid, Eisai, GlaxoSmithKline, Linde, MAP, Medtronic, Menarini, Merck, Minster, Pfizer, and St Jude, and independent support from NWO, NIH, European Community FP6, Biomed EC, the Dutch Heart and Brain Foundations, and LUMC. His spouse owns stock in Merck.
MF has, in the last 3 years, received grants and honoraria for lectures, Advisory Board membership and participation in clinical trials from Novartis, Bayer, CoLucid, Biogen-Idec, Sanofi-Aventis, Leiras, Berlin-Chemie-Menarini, Jansen-Cilag, Merck, MSD and GSK.
UR has, in the past 3 years, received grants and consultancy or industry support from Addex Pharmaceuticals, Allergan, Almirall CoLucid, Johnson & Johnson, and Vasopharm GmbH and independent support from BMBF.
AP is a consultant for CoLucid Pharmaceuticals Inc. and holds stock options in the company.
CD is a consultant for CoLucid Pharmaceuticals, Inc.
MK is a partner in FGK Clinical Research GmbH.
H-CD received honoraria for participation in clinical trials, contribution to advisory boards or oral presentations from: Addex Pharma, Allergan, Almirall, AstraZeneca, Bayer Vital, Berlin Chemie, CoLucid, Böhringer Ingelheim, Bristol-Myers Squibb, GlaxoSmithKline, Grünenthal, Janssen-Cilag, Lilly, La Roche, 3M Medica, Minster, MSD, Novartis, Johnson & Johnson, Pierre Fabre, Pfizer, Schaper and Brümmer, SanofiAventis, Weber & Weber. Financial support for research projects was provided by Allergan, Almirall, AstraZeneca, Bayer, GSK, Janssen-Cilag, Pfizer. Headache research at the Department of Neurology in Essen is supported by the German Research Council (DFG), the German Ministry of Education and Research (BMBF) and the European Union. H-CD has no ownership interest and does not own stocks of any pharmaceutical company.
Ethics committees
Germany: Essen University Hospital, Medical Faculty of the University of Duisburg-Essen, Office of the Ethics Committee. Finland: Hospital District of Helsinki and Uusimaa, Ethics Committee for Obstetrics and Gynaecology, Otorhinolaryngology, Neurology and Neurosurgery. The Netherlands: Leiden University Medical Centre, Committee Medical Ethics.
Footnotes
Acknowledgements
This study was sponsored by CoLucid Pharmaceuticals, Inc., and conducted on their behalf by FGK Clinical Research GmbH.
The study was designed by MF, MDF, AJP, CD and H-CD. MF, MDF and H-CD were principal investigators for their respective countries. UR treated subjects in the study and participated in data interpretation. CD and MK were responsible for the statistical analysis. AJP and MDF wrote the initial drafts of the manuscript which was reviewed and intensively revised by all authors. All authors had access to the full analysis.