
Abstract
A young woman had typical cluster headache attacks and a pituitary mass lesion. The headache attacks resolved after transsphenoidal resection of the tumour, which was diagnosed as a granulomatous inflammation. The association between cluster headache and granulomatous enlargement of the pituitary gland has never been described before. This case reinforces the growing evidence that even in typical cases of cluster headache, neuroimaging is mandatory to exclude structural lesions.
Introduction
Cluster headache (CH) is a severe primary headache disorder characterized by short-lasting headache attacks accompanied by ipsilateral autonomic phenomena (1). The exact pathogenesis is unknown, although vascular and hypothalamic mechanisms are proven to be involved (2). According to the criteria of the International Headache Society the diagnosis of CH is not allowed in the case of an underlying disorder (1). However, many structural lesions causing CH-like syndromes have been reported (3). Here, we describe a young woman with typical CH caused by a pituitary tumour, with a surprising histological finding.
Case report
A 26-year-old woman had a sudden onset of severe headache attacks 4 months before admission. The attacks lasted 2 to 3 h, with a frequency up to six per day. The pain was strictly localized at the right side, mostly at the right temple, but sometimes also at the right nasal cavity, eye or ear. During the attacks there was ipsilateral tearing and once there was a miosis of the right pupil and a swelling of the right eyelid. The patient reported occasional blurred and grey vision in the right eye. During attacks she was restless.
Her medical history was uneventful. Her mother suffered from migraine. Neurological examination was normal apart from a slightly smaller right pupil. A diagnosis of cluster headache was made. Both oxygen (100%, 7 l/min for 15 min) and sumatriptan (20 mg nasal spray) were effective in treating attacks and verapamil (480 mg/day) clearly reduced the attack frequency and severity. Four months later, however, attack frequency increased despite increasing the dose of verapamil up tot 720 mg/day. A cerebral MRI scan revealed a mass lesion in the sella with suprasellar extension; there was no invasion of the cavernous sinus (Fig. 1a, b). Prolactin level was normal (0.63 IU/l, normal range 0.10–0.78 IU/l). The thyreotropic axis dropped out and the patient was substituted with levothyroxin. An initial diagnosis of non-functioning adenoma was made. Several weeks/months later, galactorrea and hyperprolactinaemia developed, probably caused by compression of the pituitary stalk or by verapamil. As a trial, the patient was treated with cabergoline (0.5 mg once a week), a dopamine agonist, which had no effect on the headache attacks. Seven months later she developed bitemporal hemianopsia and a transsphenoidal resection of the tumour was performed. After resection, a diabetes insipidus developed, which was treated with desmopressin. Postoperatively, there was a dramatic reduction in attack frequency and severity. Verapamil could first be reduced to 200 mg/day, leaving only infrequent attacks of only small intensity, and later be stopped completely.
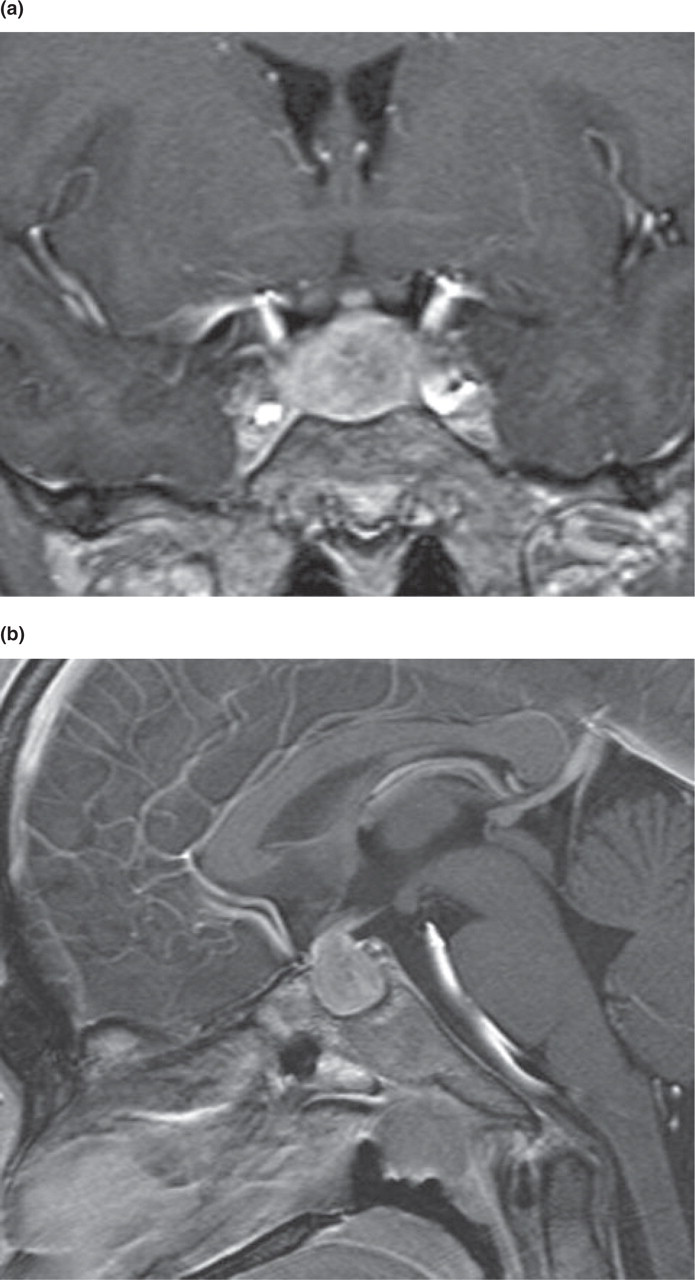
Gadolinium enhanced T1 weighted coronal (a) and sagittal (b) MRI showing the mass lesion in the sella.
Histology of the pituitary tumour showed a non-caseating granulomatous inflammation with multinucleate giant cells with Schaumann bodies (Fig. 2). An X-ray of the chest showed no signs of sarcoidosis and a Mantoux test was negative. A diagnosis of idiopathic granulomatous hypophisitis was made. Follow-up for 1 year revealed no relapse of headache.

Tissue sample of the pituitary mass lesion: non-caseating granulomatous inflammation with multinucleate giant cells with Schaumann bodies.
Discussion
Our patient had a typical primary chronic CH, fulfilling the criteria of the IHS (1). Initially, she also showed an excellent response to standard CH treatment. Because of a sudden progression and loss of efficacy of treatment, a cerebral MRI was performed, which revealed a pituitary mass lesion. After surgical removal of the lesion the headache attacks virtually disappeared, strongly suggesting a causal relation between the structural abnormality and the headache attacks.
This case adds to a growing number of patients in whom a structural lesion proved responsible for typical CH attacks (3). Most lesions were found in the region of the sella, in most cases a prolactinoma. Only one patient had a non-functioning adenoma, suggesting that tumour activity may be important. A granulomatous pituitary mass lesion has not been reported before as a cause of CH attacks. A recent study also showed that headache is a common symptom of pituitary disease, although no clarity was given about the causality between the tumour and the headache in individual patients (4).
Granulomatous involvement of the pituitary can occur in Wegener's granulomatosis, sarcoidosis, idiopathic granulomatous hypophysitis and infectious diseases with formation of granulomas such as tuberculosis. A definite diagnosis is often difficult (5). Most of these patients present with hypopituitarism, diabetes insipidus, hyperprolactinaemia, meningitis, visual disturbance due to mass effect, or a combination of the above. Presentation with headache is rare and none presented with CH. This case report stresses the importance of neuroimaging in patients with even typical CH.