
Abstract
The purpose of this study was to use intravital microscopy to determine the effect of a selective adenosine A1 receptor agonist, GR79236 (1, 3 and 10 μg/kg i.v.), on neurogenic dural blood vessel dilation in anaesthetized rats. Vasodilation was evoked either by electrical stimulation of perivascular trigeminal nerves or by intravenous CGRP. GR79236 (1-10 μg/kg i.v.) caused a dose-dependent inhibition of neurogenic vasodilation, but had no significant effect on dural vasodilation caused by CGRP. GR79236 (1-3 μg/kg i.v.) had no effect on basal dural vessel diameter, but caused transient dose-dependant bradycardia and hypotension. Bradycardia was more prolonged following 10 μg/kg i.v. GR79236. Pre-treatment with the adenosine A1 receptor antagonist DPCPX (1 μg/kg i.v.) prevented the inhibitory effect of GR79236 (10 μg/kg i.v.) on neurogenic vasodilation as well as GR79236-induced bradycardia and hypotension. These data suggest that the inhibition of neurogenic vasodilation by GR79236 is mediated via the activation of prejunctional adenosine A1 receptors. Provided the systemic cardiovascular effects could be limited, such a mechanism may offer a novel approach to migraine therapy.
Introduction
Adenosine is an endogenous neuromodulator that exerts effects on many cells, mainly by the activation of either low affinity (A2B and A3) or high affinity (A1 and A2A) adenosine receptors. The modulatory role of adenosine within the nervous system was recognized several years ago when Hedqvist and Fredholm (1) showed that α-adrenoceptor-mediated vasoconstrictor responses could be potentiated by adenosine. Relatively recently, it has also been found that adenosine, acting via adenosine A1 receptors within the spinal cord, is involved in the modulation of both acute and chronic pain states in rats (2). This may have useful therapeutic potential as many patients experiencing neuropathic pain have a deficiency in circulating adenosine and, when infused at low doses, adenosine can alleviate ongoing neuropathic pain (3). The trigeminal nerve conveys sensory information from the head to the CNS and activation of these nerves is thought to be one of the key factors underlying the generation of migraine headache pain (4). Adenosine A1 receptors have recently been found on cell bodies in the trigeminal ganglia of humans (5) and rats (Schindler, unpublished observations).
In order to probe the function of the adenosine A1 receptor in the trigemino-vascular system we have used intravital microscopy to determine the effect of the highly selective A1 receptor agonist, GR79236 (N-[(1S, trans)-2-hydroxycyclopentyl]adenosine (6) on trigeminally evoked dural blood vessel dilation or by administration of exogenous calcitonin gene-related peptide (CGRP) as the vasodilator response to electrical stimulation of the trigeminal nerve has previously been shown to be mediated by this peptide (7). We have also studied the effects of GR79236 on electrically evoked dural vasodilation in the presence of the selective adenosine A1 receptor antagonist, 8-cyclopentyl-1,3-dipropylxanthine (DPCPX). Some of the data presented here have been published in preliminary form at Headache World 2000 (8).
Methods
Surgical procedures
Male Sprague-Dawley rats were anaesthetized using pentobarbitone (65 mg/kg i.p.). Both femoral veins were cannulated for drug administration and maintenance of anaesthesia (18 mg/kg/h pentobarbitone). The left femoral artery was cannulated for continuous measurement of blood pressure and heart rate. Rectal temperature was monitored and maintained at 37–38°C with a heated blanket and thermistor. The procedure for intravital microscopy experiments was essentially as described previously (7). Briefly, rats were placed in a stereotaxic frame and the parietal bone was thinned using a saline-cooled drill until the middle meningeal artery (MMA) became visible. The area was covered with warm mineral oil to prevent drying and the preparation was left for up to an hour for the vessel diameter to normalize. The artery was viewed under green filtered light using an intravital microscope (Moritex Europe Ltd) attached to a computer equipped with a video capture card (Hauppauge, London, UK). Dural blood vessel diameter was continuously measured using a video dimension analyser (Living Systems Instrumentation, Burlington, VT) and the data, along with the blood pressure and heart rate, were recorded using a Notocord Hem 3.3 Data Analysis system.
This study complied with United Kingdom (UK) legislation and Glaxo Smith Kline policy on the care and use of animals.
Electrode placement on the cranial window
For neurogenic vasodilation experiments, a bipolar stimulating electrode (NEX-200, Clark Electromedical Instruments, Reading, UK) was placed on the surface of the cranial window less than 200 µm from the section of vessel under study. Contact pressure was adjusted to ensure adequate transcranial stimulation whilst ensuring that blood flow was not compromized. Stimulations at 5 Hz with a 1-ms pulse width were applied for 10 s with increasing voltage until an intensity was reached at which a maximal dilation was observed. Subsequent electrically induced responses were then evoked using submaximal stimulus intensity (approximately 70–80% of maximum).
Experimental procedures
Effect of GR79236 on neurogenic dural vasodilation
A control response to electrical stimulation was obtained and repeated at 20-min intervals on two or three further occasions. Each dose of GR79236 was administered as a bolus intravenous injection via the femoral vein cannula. Rats were treated 5 min prior to each post-control stimulation with GR79236 in cumulative doses (1–30 µg/kg i.v., n=2–5) or vehicle (sterile saline, n=2–5).
Effect of GR79236 on CGRP induced dural vasodilation
A control injection of rat αCGRP (0.3 µg/kg i.v.) was administered, followed by two further injections at 20-min intervals. Rats were treated with GR79236 (10 µg/kg i.v., n=5), or vehicle (sterile saline, n=5), 5 min prior to the second dose of CGRP.
Effect of DPCPX and GR79236 on neurogenic dural vasodilation
Rats were pre-treated with the selective A1 receptor antagonist DPCPX (1 mg/kg i.v., n=5), or vehicle (n=5), 5 min before a control electrical stimulation was made. This stimulation was repeated on two further occasions at 20-min intervals. Rats were treated with GR79236 (10 µg/kg i.v.) 5 min prior to the second electrical stimulation.
Data analysis
The effects of CGRP or electrical stimulation on dural vessel diameter were calculated as the maximum percentage increase from pre-dose or pre-stimulation baseline values. Data are presented as mean ± standard error of the mean. The effect of GR79236 on dural vessel dilation was assessed by comparing the control responses elicited by electrical stimulation with the responses made in the presence of test drug. Dural vessel diameter was measured in arbitrary units throughout these studies. Statistics were performed using
Drugs
All drugs were administered intravenously in a volume of 1 ml/kg and doses refer to free base weight. Rat αCGRP (Bachem, Meyerside, UK) was initially dissolved in sterile water and aliquots were frozen. Subsequent dilutions were made in 0.9% saline. GR79236 was synthesized de novo in the laboratories of GlaxoWellcome and was dissolved in sterile water with subsequent dilutions made in 0.9% saline. DPCPX (RBI, Gillingham, UK) was initially dissolved in warmed dimethylsulphoxide (DMSO; 8% v/v) and sodium hydroxide (1
Results
Effect of GR79236 on electrically induced dural vasodilation
Electrical stimulation of the cranial window initially caused a transient vasoconstriction lasting between 10 and 15 s followed by a dilation which persisted for up to 5 min. The maximum increase in vessel diameter observed was approximately 170–180%. Responses were reproducible for three to four stimulations, after which the vessels did not always fully return to baseline values. GR79236 had no effect on resting vessel diameter at doses up to 10 µg/kg. Higher doses were not used as they caused a long-lasting vasodilation that persisted for over 1 h. GR79236 caused a dose-dependent inhibition of neurogenic vasodilation following 1 µg/kg i.v. (8.9 ± 1.5%, n=2), 3 µg/kg i.v. (19.1 ± 5.2%, n=5) and 10 µg/kg i.v. (54.3 ± 6.2%; P<0.01, n=5) compared with the pre-treatment control stimulation (Fig. 1).

The dose-related inhibition of electrically evoked MMA vasodilation following GR79236. Data are plotted as the percentage reduction of response compared with the pre-treatment control stimulation. Values are means ± standard error of the mean. Statistical analysis was performed by
Effect of GR79236 on CGRP-induced dural vasodilation
Intravenous injection of rat αCGRP caused dose-related increases in dural artery diameter (ED50: 0.28 ± 0.07 µg/kg, unpublished data). The dose of rat αCGRP used in the current study (0.3 µg/kg i.v.) was chosen as it caused submaximal increases in dural artery diameter that were highly reproducible in vehicle-treated animals. The control response to rat αCGRP (59.8 ± 11.6% increase in diameter, n=5) was not significantly inhibited following GR79236 (10 µg/kg i.v.) after 5 min (66.2 ± 13.7%) or 20 min (58.7 ± 12.7%) (Fig. 2).
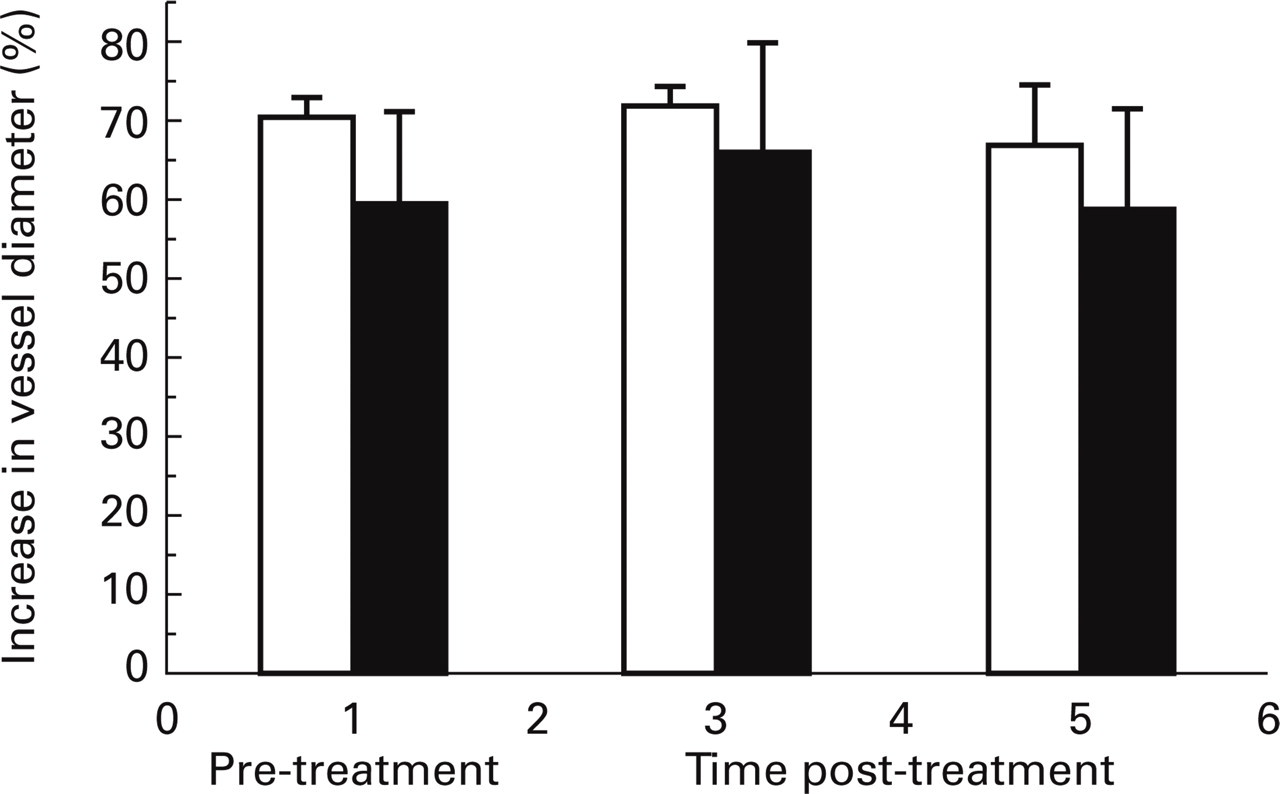
The effect of GR79236 (10 µg/kg i.v.; closed bars) or vehicle (saline; open bars) treatment on increases in dural vessel diameter following rat α-CGRP (300 ng/kg i.v.). Data are mean percentage increases in vessel diameter ± standard error of the mean, n=5.
Effect of DPCPX and GR79236 on neurogenic dural vasodilation
DPCPX (1 mg/kg i.v.) or vehicle administration had no effect on dural vessel diameter per se. Pre-treatment with DPCPX (1 mg/kg i.v.) prevented the inhibitory effect of GR79236 (10 µg/kg i.v.; Fig. 3).

Effect of GR79236 (10 µg/kg i.v.) treatment on electrically evoked increases in dural vessel diameter in animals previously dosed with DPCPX (1 mg/kg i.v.; open bars) or DPCPX vehicle (closed bars). Data are mean percentage increases in vessel diameter ± standard error of the mean. Statistical analysis was performed by
Cardiovascular effects of GR79236 and DPCPX
Before treatment with GR79236, animals had a baseline resting mean arterial blood pressure (MABP) of 95 ± 1 mmHg and a mean resting heart rate of 267 ± 20 beats per minute (n=6). Intravenous injection of GR79236 (1–10 µg/kg) caused transient dose-related hypotension and bradycardia (Fig. 4). Blood pressure always returned to pre-treatment levels before electrical stimulation was commenced and, following the lower doses of GR79236 (1–3 µg/kg), heart rate also returned to a steady baseline within 5 min. Following the highest dose of drug (10 µg/kg), the post-treatment heart rate baseline remained lower than its pre-treatment level (− 10.7 ± 1.7%). Intravenous injection of DPCPX (1 mg/kg i.v.) had no effect on heart rate and caused only a small (<10 mmHg) and transient increase in mean arterial blood pressure followed by a small decrease before the baseline was restored (after approximately 1 min). Vehicle administration had no effect on these parameters. DPCPX treatment abolished the effect of GR79236 on blood pressure and heart rate.

The effect of GR79236 on mean arterial blood pressure (solid line) and heart rate (dashed line). Data are the maximum percentage inhibition of the mean resting blood pressure (95 ± 1 mmHg) and a mean resting heart rate (267 ± 20 bpm) immediately following the administration of GR79236 (1–10 µg/kg i.v.). Blood pressure always returned to pre-treatment levels before electrical stimulation was commenced and, following the lower doses of GR79236 (1–3 µg/kg), heart rate also returned to a steady baseline within 5 min. Following the highest dose of drug (10 µg/kg), the post-treatment baseline remained lower than its pre-treatment level (− 10.7 ± 1.7%).
Discussion
Intravenous administration of CGRP, substance P and neurokinin A to rats causes dose-related increases in dural artery diameter, presumably by a post-junctional action on MMA smooth muscle cells (7). In the present study we have shown that transcranial electrical stimulation of perivascular trigeminal nerves causes a transient vasoconstriction, probably due to release of catecholamines from sympathetic nerve stimulation, followed by a marked and reproducible vasodilation of the dural vasculature. This vasodilator response is thought to be mediated by CGRP release from activated Aδ sensory afferent fibres in the dura mater, as it is inhibited by the CGRP antagonist, human-αCGRP8-37 (10). Pre-treatment with the rat selective NK1 antagonist RP67580, however, has no effect on the response (10), suggesting that substance P is not involved. These observations have been supported by results from our own laboratory, which show that the selective NK1 antagonist GR205171 had no effect on electrically evoked vasodilation (unpublished observations) in doses that abolished increases in dural plasma protein extravasation in response to electrical stimulation of the trigeminal ganglion (9).
The results from the present study also indicate that electrically evoked dural vasodilation may be dose-dependently reduced by the prior administration of the selective adenosine A1 agonist, GR79236, at doses up to 10 µg/kg (see [6] for selectivity profile). However, dural vasodilation evoked by CGRP (0.3 µg/kg) was not inhibited by pre-treating the animal with GR79236. This dose of CGRP was chosen as it was close to the previously derived ED50 for this peptide (0.28 ± 0.07 µg/kg; unpublished data). Hence we would conclude that the inhibition of electrically evoked dilation by GR79236 is neither by a post-junctional antagonism of the CGRP effect nor by acting as a functional antagonist on the vessel wall, but would appear to be via a pre-junctional inhibition of CGRP release from perivascular trigeminal sensory neurones.
Doses higher than 10 µg/kg GR79236 caused a long lasting vasodilation of the dural vessels per se and therefore were not used in this study. GR79236 also caused small dose-dependent decreases in blood pressure and heart rate. Blood pressure always returned to pre-dose baseline values before the electrical stimulation was started, but following 10 µg/kg GR79236 the bradycardia was maintained at a lower baseline, approximately 10.7% below the pre-dose baseline. This maintained bradycardia is presumably due to a direct effect of GR79236 on cardiac A1 receptors and reflects the duration of action of this compound.
We have also used the selective adenosine A1 antagonist DPCPX in this study to confirm that the effects of GR79236 are mediated by an A1 receptor mechanism. This compound has been reported to be over 700-fold selective for adenosine A1 receptors vs. adenosine A2A and A2B receptors in rat brain slices (11). Intravenous injection of DPCPX alone did not markedly alter blood pressure, heart rate or dural vessel diameter, suggesting that there is no endogenous adenosine release acting to decrease the resting vessel tone in these animals. In the presence of DPCPX, electrically evoked dural vasodilation was not inhibited by GR79236 (10 µg/kg i.v.) when compared with the control stimulation (also carried out in the presence of DPCPX), and the previously observed bradycardia and transient hypotension were abolished. Following DPCPX vehicle administration, however, as expected, vasodilation was significantly inhibited by GR79236 (53.4 ± 11.9%). This inhibition is not significantly different from the percentage inhibition of vasodilation that occurred following 10 µg/kg GR79236 in the initial dose–response study (54.3 ± 6.2%; Fig 1).
Taken together these results indicate that GR79236 acts at adenosine A1 receptors on the peripheral terminals of perivascular afferent sensory neurones, resulting in an inhibition of neurogenic dural vasodilation. Although cardiovascular side-effects (decrease in blood pressure, bradycardia) were observed after systemic administration of GR79236 and may limit the utility of this compound in the clinic, we believe that these effects are unlikely to account for the inhibition of neurogenic vasodilation. Indeed, blood pressure had always returned to baseline levels before stimulation of neurogenic vasodilation. In addition, GR79236 had no effect on CGRP-evoked vasodilation whilst still producing the same direct effect on heart rate and blood pressure. It is also clear that these direct effects on blood pressure and heart rate, as well as the inhibitory effect on dural vasodilation, were mediated by an A1 receptor mechanism as they were abolished by pre-treatment with the adenosine A1 antagonist DPCPX.
Dural blood vessel dilation in the rat evoked by transcranial electrical stimulation under similar conditions to those used in the present study is significantly inhibited by sumatriptan (7) and naratriptan (ED50: 3.1 ± 0.9 mg/kg i.v., unpublished data). This inhibition is thought to be due to inhibition of CGRP via activation of pre-junctional trigeminal fibres (7). Studies of cranial venous neuropeptide levels have shown that during migraine attacks CGRP, but not SP, is elevated in the external jugular vein blood in both adolescents (12) and adults (13) and that these levels are normalized following sumatriptan administration (14). This suggests that the therapeutic effect of sumatriptan may at least in part be mediated through an inhibition of the release of CGRP. As the present intravital microscopy study provides evidence for a similar mechanism of action with GR79236, it is tempting to speculate that if this activity could be dissociated from the effects on blood pressure and heart rate, then adenosine A1 agonists would also be effective anti-migraine agents. Furthermore, as adenosine A1 agonists also inhibit the firing of second order neurones within the trigeminal caudalis in response to activation of trigeminal afferents (15), it is possible that they may be clinically effective in migraine as a consequence of an inhibitory effect both in the brain as well as the periphery.