
Abstract
Familial cluster headache (CH) was analysed in 21 Swedish families. Diagnosis was made according to The International Classification of Headache Disorders 2004. We identified 55 affected, of whom 42 had episodic or chronic CH, one had probable CH and 12 had atypical symptoms. The atypical cases did not fulfil the diagnostic criteria for CH, but had clinical symptoms with more resemblance to CH than to migraine or other trigeminal autonomic cephalgia syndromes. The overall male : female ratio was 1.8 : 1. The overall mean age at onset was significantly lower in the second/third generation than in the first generation (mean age at onset 22 vs. 31 years, SD ± 7 vs. 13 years; P < 0.01). This may be anticipation or selection bias, since individuals with late age at onset from the second/third generation may not yet have symptoms. The prevalence of migraine was 24% (13/55), i.e. similar to the prevalence in the general population. The high incidence of atypical CH cases in the Swedish families with other members affected with CH may suggest that the spectrum of CH is broader than previously thought. We suggest that atypical CH in CH families may represent an expanded spectrum of the disease with a common aetiology, i.e. a common genetic background.
Introduction
Cluster headache (CH) is a primary headache disorder characterized by attacks of severe or very severe strictly unilateral pain, ipsilateral autonomic features and/or restlessness or agitation [Headache Classification Subcommittee of the International Headache Society (IHS) (1)]. The periodicity of attacks inspired to its name (2–4). CH is considered relatively rare with a prevalence of 1 in 1000–1500 in the general population (5, 6). However, recent epidemiological surveys indicate that the prevalence is more likely to be about 1 : 500 (7, 8). The majority suffer from episodic CH, but about 10% have chronic CH (9, 10). CH is more common in men than in women, although the male : female ratio has decreased every decade from about 6 : 1 before the 1960s to 2–3 : 1 in the 1990s (10–12).
The aetiology and pathophysiology in CH are not yet completely understood, but are presumed to involve an activation of hypothalamic and trigeminovascular systems (13). Functional imaging with positron emission tomography (PET) shows activation of the ipsilateral hypothalamic grey area during attacks of CH compared with the headache-free state (14). This activation has not been observed in migraine, experimental pain induced by capsaicin injection to the forehead or in CH patients not experiencing attacks after nitroglycerine administration (15–17). Although PET shows specific local brain activation in CH, the diagnosis of CH still relies exclusively on the headache history. The International Classification of Headache Disorders is based on explicit diagnostic criteria, i.e. ‘unambiguous, precise and with as little room for interpretation as possible’ (1). The 2nd edition of The International Classification of Headache Disorders classifies CH among the newly described Trigeminal Autonomic Cephalgias (TAC) (18) and probable CH is introduced as attacks fulfilling all but one of criteria A–D for CH (1). Patients with one cluster period only should be diagnosed cluster headache 3.1 in the new classification, in contrast to the former classification when two periods or continuous attacks without remission were needed for a CH diagnosis (1, 19, 20). However, the clinical spectrum of CH may be even broader. For example, CH sine headache has been reported but is not included in the classification (21, 22).
It may be difficult to judge and delineate the clinical spectrum from sporadic cases of CH only. About 5–10% of CH patients have familial CH (23–28). An analysis of familial cases may provide more insight into the clinical spectrum of phenotypes of CH. A correct classification of whether members of families are affected or non-affected is of great importance in genetic epidemiological and molecular genetic studies. We have collected clinical information from members of CH families reported to have two or more affected with CH with the aim of determining CH affection status. In this study we present the analysis of phenotypes of CH in 21 Swedish CH families.
Patients and methods
CH has been studied for almost four decades at our Department of Neurology; initially at Karolinska Hospital and Söder Hospital in Stockholm, and since 1997 at Karolinska University Hospital, Huddinge, at present comprising 604 CH patients (12). Probands with CH who reported one or more possibly affected relatives with CH were consecutively collected from this group of patients. Twelve affected subjects belonged to five families. We received eight families from headache specialists from other departments and clinics. Another two families were identified via an advertisement on Svenska Migränförbundet, website http://www.migran.org, a network for headache patients in Sweden. Probands with a supposed familial occurrence of CH were contacted by mail and/or by telephone, and were informed about the study. The probands provided names and addresses of possibly affected relatives and non-affected first- or second-degree relatives who would be willing to participate. They all received a questionnaire regarding CH and other types of headaches and were also asked to report details of affected relatives, like the probands. Those not responding were contacted a second time by telephone or mail. All possibly affected were personally interviewed by a neurologist specialized in headache (C.S.). Only those with clinical documentation (through case records, questionnaire and/or personal interview) were included in the analyses. The 2nd edition of The International Classification of Headache Disorders was used for headache diagnosis with respect to CH, other TACs and migraine (1). If two or more of the CH criteria (A–E) were not fulfilled patients were classified atypical CH, if the headache could not clearly be classified as migraine, tension-type headache or another type of headache described in the classification. Only families with at least two cases of suspected CH were included in the study, as we considered CH to be familial if this criterion was met. The study was approved by the Local Ethics Committee.
Data management and statistical analyses
Retrospectively ranked worst pain intensity during an attack was scored on a pain scale from 0 to 10. These numbers were transformed to pain characteristics of the IHS classification as follows; 0 = no pain, 1–3 = mild pain, 4–6 = moderate pain, 7–9 = severe pain and 10 = very severe pain.
Statistical analyses were performed using GraphPad InStat version 3.00 for Windows 95 (GraphPad Software, San Diego CA, USA, http://www.graphpad.com). Wilcoxon match-pairs signed rank test, Fisher's exact test and standard deviation were used. All tests of significance were two-tailed using 5% significance level.
Results
Thirty families with at least two cases of suspected CH were identified. Nine families were excluded because only one affected was still alive in six families, two families declined to participate and in one family it was impossible to reach the affected relative. A total of 21 families were available for evaluation. Figure 1 shows pedigrees of families including atypical cases, and pedigrees of all 21 families are shown in Appendix 1 which is available online. Four deceased with possible CH were labelled affected in the pedigrees (families 9, 12 and 14), but they were not included in our analyses, since their diagnoses were not verified by clinical documentation. One person with CH was not included in the analysis, since the clinical record could not be found and relatives regarded her as being too old for an interview (family 9). All families were of Caucasian origin except for family 14, which was of Afro-American origin, except for the non-affected Caucasian mother in the second generation.

Pedigrees of families with atypical cluster headache cases.
Probands and affected relatives in familial CH
We identified 42 subjects with CH (27 men and 15 women) in 21 families. One subject had probable CH. Twelve subjects (seven men and five women) from nine families had clinical symptoms with resemblance to CH, but they did not fulfil all the diagnostic criteria for CH. They were classified as atypical CH (Tables 2, 3 and 4). Ten cases had been reported by the probands to have CH similar to their own CH. One proband was aware of a relative with headache of unknown character and one was recruited as a non-affected relative by the proband, but atypical CH was revealed during the interview. We also received information from 31 first- and second-degree relatives unaffected by CH (13 men and 18 women, mean age 60 years, range 14–86 years) by personal telephone interview. Thus we personally interviewed 86 subjects (55 sufferers from typical, probable or atypical CH and 31 non-affected).
Gender ratio
The male : female ratio was 1.9 : 1 in CH and 1.4 : 1 in atypical CH, while the combined male : female ratio was 1.8 : 1. The difference between gender ratio in typical and atypical CH was non-significant (P = 0.74).
Age at onset
The mean age at onset was similar among those with CH and atypical CH (27 years, range 12–56 years, SD ± 11) vs. 27 years (range 12–58 years, SD ± 12, P = 0.76). In those with CH the mean age at onset was 30 years (n = 24) in the first generation and 22 years in the second/third generation (n = 14 and n = 4), respectively. In those with atypical CH the mean age at onset in the first generation (n = 5) was 33 years and in the second/third generation 24 years (n = 6 and n = 1). The combined mean age at onset in CH and atypical CH was significantly lower in the second and third generations than in the first generation (31 vs. 22 years; P < 0.01).
Clinical characteristics of atypical cluster headache
Table 1 shows demographic data, Tables 2 and 3 the clinical characteristics and Table 4 the 12 atypical CH cases in relation to a CH diagnosis by International Classification of Headache Disorders (1). They all had strictly unilateral pain in the orbital/periorbital area, but in two persons the side of pain could change. The intensity of worst pain during attacks was very severe or severe in all cases except one. All cases except one had one or more ipsilateral autonomic symptoms during attacks. Ten out of 12 patients also reported restlessness and/or agitation. However, the temporal pattern of headaches was different and did not agree with the IHS criteria either with respect to duration of attacks or frequency of attacks. Rather than having clusters of headache attacks, the attacks occurred in a sporadic pattern in 11 of 12 individuals. Nine out of 12 had attacks that were longer than 3 h. The difference of the temporal pattern along with one other missing feature was the reason why they could not be classified as either CH or probable CH.
Demographic data on atypical cluster headache

Pain characteristics of atypical cluster headache
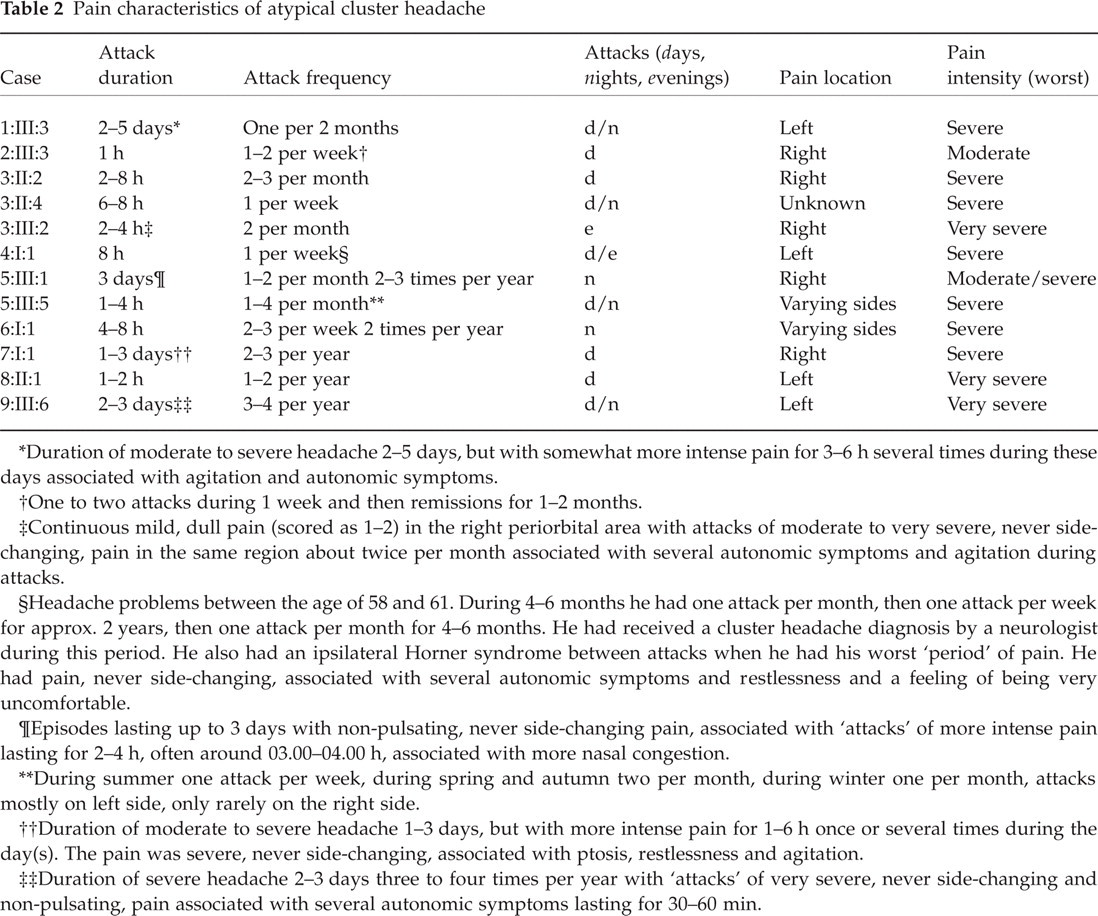
Duration of moderate to severe headache 2–5 days, but with somewhat more intense pain for 3–6 h several times during these days associated with agitation and autonomic symptoms.
One to two attacks during 1 week and then remissions for 1–2 months.
Continuous mild, dull pain (scored as 1–2) in the right periorbital area with attacks of moderate to very severe, never side-changing, pain in the same region about twice per month associated with several autonomic symptoms and agitation during attacks.
Headache problems between the age of 58 and 61. During 4–6 months he had one attack per month, then one attack per week for approx. 2 years, then one attack per month for 4–6 months. He had received a cluster headache diagnosis by a neurologist during this period. He also had an ipsilateral Horner syndrome between attacks when he had his worst ‘period’ of pain. He had pain, never side-changing, associated with several autonomic symptoms and restlessness and a feeling of being very uncomfortable.
Episodes lasting up to 3 days with non-pulsating, never side-changing pain, associated with ‘attacks’ of more intense pain lasting for 2–4 h, often around 03.00–04.00 h, associated with more nasal congestion.
During summer one attack per week, during spring and autumn two per month, during winter one per month, attacks mostly on left side, only rarely on the right side.
Duration of moderate to severe headache 1–3 days, but with more intense pain for 1–6 h once or several times during the day(s). The pain was severe, never side-changing, associated with ptosis, restlessness and agitation.
Duration of severe headache 2–3 days three to four times per year with ‘attacks’ of very severe, never side-changing and non-pulsating, pain associated with several autonomic symptoms lasting for 30–60 min.
Associated symptoms of atypical cluster headache

Atypical cluster headache (CH) cases in relation to the International Headache Classification of Headache Disorders

Four subjects described moderate to severe pain for several days, which was aggravated one or more times during the period (1:III:3, 5:III:1, 7:I:1 and 9:III:6). One person (3:III:2) had continuous mild pain in the orbital/periorbital area with infrequent attacks of more intensive pain in the same area. Subject 3:II:2 had sporadic attacks of severe unilateral, non-pulsating pain lasting for 2–8 h associated with several autonomic symptoms and agitation. All subjects except one (8:II:1) reported one or more local, ipsilateral autonomic symptoms during attacks. This latter subject described her attacks as being similar to her mother's CH, except that she had only sporadic attacks and lacked autonomic symptoms. Five were restless and eight were agitated during attacks. Only two of the 12 atypical CH cases were neither restless nor agitated during attacks; one of them had autonomic symptoms, while the other had no autonomic symptoms. Pain was aggravated by alcohol in five of the subjects. Ten of 12 reported nausea and nine of 12 also had increased sensitivity to light. Further clinical description and comments on several separate cases are shown in Table 2.
The mean age of the atypical CH sufferers was 50 years (range 21–75 years) and the mean duration of disease was 25 years (4–45 years).
Clinical characteristics of familial CH
Among the 42 CH cases, 39 had episodic CH and three had chronic CH. The mean duration of episodic cluster periods was 6.4 weeks (n = 37). Two persons could not describe the exact duration of periods of their episodic CH.
The mean attack frequency per day during active periods of CH was 3.2 (SD ±2, n = 42). However, all affected in families 6, 13 and 21 reported, interestingly, a high daily attack frequency; the two with typical CH in family 6 had 6–8 and 2–6 attacks per day, the three affected in family 13 had 5–8, 4 and 1–6 attacks per day, respectively, and the two affected in family 21 had 5 and 3–8 attacks per day. Of these three families only family 6 had a member affected with atypical CH.
All patients with typical CH described one or more associated autonomic symptom on the pain side during attacks; 35 (83%) had conjunctival injection, 37 (88%) had lacrimation and 33 (79%) had nasal congestion and/or rhinorrhoea. Fifteen (36%) described miosis on the affected side, but 20 persons were uncertain about miosis. Twenty-eight (67%) described ptosis, but seven persons did not know if they had ptosis during attacks.
Twenty-seven (64%) described increased sensitivity to light and 15 (36%) had nausea during attacks. Twenty-three (55%) reported provocation of attacks by alcohol.
Clinical characteristics of probable CH
One person (12:II:1) fulfilled IHS diagnostic criteria for probable CH. This subject had been suffering from one to four severe left-sided attacks per month, two ‘periods’ lasting for 3–4 weeks to 2–3 months per year, since the age of 16. The attacks had a duration of 2 h and were associated with ipsilateral conjunctival injection, lacrimation, nasal congestion, restlessness and agitation.
Pain characteristics
All CH sufferers had strictly unilateral pain. Six had unilateral pain with side changing location; four had a side of pain that was more dominant. The change of side occurred within cluster periods in two subjects and between cluster periods in the remaining four. Two of those with side-changing symptoms belonged to the same family as well as another subject with atypical CH without side-changing symptoms.
Nine of the 21 families (including CH, probable CH and atypical CH) described symptom location on the same side in all affected subjects within the family; six families had left-sided symptoms only and three families had right-sided symptoms. Thirty-five of the 42 with typical CH described their worst pain during attacks as being very severe, while the remaining seven had severe pain intensity.
Migraine
Twenty-four of the 86 interviewed subjects (28%) had migraine according to the International Classification of Headache Disorders 2004 (1). Twenty-one had migraine without aura and three had migraine with aura. Nine of the 42 with CH also had migraine (21%), seven had migraine without aura and two had migraine with aura. The subject with probable CH had no migraine. Four with atypical CH had migraine (33%), of whom three had migraine without aura and one had migraine with aura. Altogether, 13 out of 55 (24%) with typical or atypical CH had migraine. One person (14:II:1) had visual aura followed by severe, throbbing, unilateral pain which was aggravated by movement, accompanied by nausea and vomiting and associated with ipsilateral conjunctival injection during attacks. The attacks lasted 8–12 h and he had to rest in a dark and silent room. Due to the characteristic pain description he was diagnosed as migraine with aura with associated autonomic symptoms, while his deceased mother was reported to have CH and his two sons had CH. Eleven of the 31 non-affected relatives (35%) had migraine without aura.
Discussion
During recent years it has been shown that CH is an inherited disorder in some families. Publications from 1947 to 1985 reported that 47 first-degree relatives were affected in 1182 families (29). This suggests an increased family risk even though the suspected diagnosis was not confirmed in all families. Recent genetic epidemiological surveys provide more complete information about the relatives (23–28, 30). First-degree relatives were found to have a 5–18-fold significantly increased risk of CH compared with the general population, while second-degree relatives had a one- to three-fold significantly increased risk (30). Theoretically, a shared environment can produce relative risks of the magnitude observed for CH only under extreme conditions (31). CH has been reported in seven concordant monozygotic twin pairs (32–38). This also indicates the importance of genetic factors, although publication itself introduces selection bias (39). The latter is emphasized by the only published large twin survey based on the Swedish Twin Registry and the Swedish Inpatient Registry (40). Thus, two monozygotic and nine dizygotic twin pairs were all discordant for CH and had been discordant between 10 and 31 years.
We analysed the clinical spectrum of familial CH in greater detail and were able to identify 12 subjects with headache similar to CH, atypical CH, belonging to nine CH families. They all had clinical CH characteristics, but the attacks occurred in a sporadic way rather than in clusters, although some had a tendency towards ‘mini-bouts’ (41). Some had pain described to be somewhat milder than in typical CH. A few described very severe pain for several hours. Some reported episodes with moderate to severe headache lasting for some days, but less than a week, with more intense pain for several hours during these days. One subject had continuous mild and dull pain in the orbital/periorbital area with infrequent attacks of moderate to very severe pain associated with autonomic symptoms in the same area. All atypical cases described strictly unilateral orbital/periorbital pain. All but one had autonomic features during attacks.
Some of the cases could in theory be classified as migraine without aura or episodic tension-type headache. However, combinations of clinical features such as ipsilateral autonomic symptoms, restlessness and agitation are not common symptoms of migraine with or without aura or tension-type headache. The long attack duration was different from paroxysmal hemicrania and short-lasting neuralgiform headache attacks with conjunctival injection and tearing (SUNCT). Our data on atypical CH indicate that the spectrum of CH may be broader than defined by the International Classification of Headache Disorders. At onset, CH attacks may be milder and more infrequent in some patients (2, 3, 35, 42, 43). We could thus assume that subjects with the shortest observation period amongst the atypical CH cases may develop a more typical CH within some years. On the other hand, most of the atypical cases were observed for many years after onset without a ‘transformation’ to a more typical form of CH. We suggest that atypical CH may represent an expanded spectrum of the disease and that the phenotypical variations of atypical and typical CH in our families may represent one disorder with the same genetic background.
Migraine features such as accompanying nausea and photophobia are common in patients with CH (44–47), while aura symptoms are rare in CH (48, 49). CH features may occur in migraine and vice versa (50–52). For that reason a plausible relationship between CH and migraine has been suggested (26, 53, 54). However, we, and others, regard CH and migraine as distinct separate headache disorders. First, CH, migraine without aura and migraine with aura all have distinct clinical features (1). Second, the prevalence of migraine among people with CH corresponds to the prevalence of migraine in the general population (25, 55–58). The prevalence data on migraine in our CH families further support this. Our affected CH sufferers, both the typical and the atypical CH cases, were clearly able to differentiate between CH and migraine with and without aura. The high prevalence of migraine (35%) among our series of relatives without CH is likely to be caused by selection bias, as a few were thought to have CH and for that reason included. Third, activation of the brain occur
Gender ratio
Those affected with familial and non-familial CH do have similar clinical symptoms (28, 59, 60). It is well known that sporadic cases of CH display a marked male preponderance, as mentioned in the Introduction. On the other hand, the ratio between men and women is as low as 1.2 : 1 in familial CH (23, 25–28, 59) and 2.5 : 1 in a recent British clinic population (10). A recent Italian study described a CH family with eight affected and a male : female ratio of 1 : 3 (61). We found a slightly higher male : female ratio than in previous family studies, but it was less than in the recent British clinic population. Obviously, the varying gender ratio in familial CH and non-familial CH deserves further investigation.
Age at onset
Our familial CH cases had a mean age at onset of 27 years, which is comparable to the findings in series of sporadic cases (11, 12). The mean age at onset was significantly lower in the second and third than in the first generation, a result similar to an analysis of 18 Danish CH families (59). This may suggest anticipation, but a selection bias cannot be excluded since individuals with late age at onset from the second and third generations have not got CH symptoms.
Pain characteristics
Interestingly, we found that all affected with CH in three of our families had a rather high frequency of attacks per day. This has also been seen in a Danish family (59), and this observation could be clinical evidence of genetic heterogeneity.
The side of the symptoms is likely to be distributed by chance, as those affected in some families had the symptoms on the same side, while in other families they had symptoms on different sides. Different sides of pain have also been described in a twin pair (35).
Comments on atypical CH
Even though there are several reports on familial CH, there is little detailed information about the clinical characteristics of these families. One study analysed 18 CH families for intra- and interfamilial variability of symptoms (59); except for a lower age at onset in children than in parents with CH and one family with high frequency of attacks (one to eight attacks/day) as mentioned above, there were no signs of genetic heterogeneity on clinical grounds. An Italian family study found a family history of CH in 44 of 220 probands (27). Twelve (21%) of the 57 affected relatives had CH with undetermined periodicity and 11/57 (19%) had CH-like disorders not fulfilling IHS criteria. The authors discussed the fact that they personally interviewed all possibly affected relatives and thus uncovered a high number of difficult-to-diagnose forms of CH, thereby increasing the incidence of CH in relatives. A French study found a high frequency of females in familial CH compared with sporadic CH cases (28). Recently, a Dutch study identified 75 CH families in 1720 CH patients comprising a total of 162 affected relatives (62). Twenty-four evidently had probable CH, mainly caused by long attack duration, i.e. duration of > 3 h. A second Italian study compared the clinical characteristics of sporadic and familial CH in 76 sex- and age-matched patients (60). Females with familial CH had a lower age at onset than females with sporadic CH, but otherwise there were no specific clinical differences.
An atypical pattern of sporadic CH has been described (63–65) and is also discussed above. Autonomic symptoms may be lacking in otherwise typical CH (66, 67). In one family (59) the proband lacked autonomic symptoms while the second-degree relative had autonomic symptoms, which in these cases may suggest a common aetiology of CH without autonomic symptoms and CH. The former classification of the IHS (19) required autonomic symptoms in order to diagnose CH. However, the new revised version of this classification, i.e. The International Classification of Headache Disorders (1), does not have this requirement if restlessness and/or agitation are present.
A pair of monozygotic twins initially described as discordant later became concordant for CH (35, 37). The cluster periods were very short in the beginning and one of the twins experienced attacks of very short duration. Later the characteristics of attacks changed to be typical episodic CH. The clinical spectrum may be even broader, as persons with CH sine headache have been reported (21, 22). One sufferer had only autonomic symptoms and no pain in the majority of attacks, but 6 years later he developed episodic CH (21). One woman experienced autonomic symptoms but no headaches, while her father had CH without autonomic symptoms and her son had episodic CH (22). After the son's CH had resolved there was a period with autonomic symptoms only in the same regular fashion as in the cluster period.
Conclusion
We have found 12 cases with atypical CH pattern among our CH families. We suggest that atypical CH represents an expanded spectrum of the disease and that phenotypical variations of typical and atypical CH may represent one disorder with the same genetic background. However, final proof requires identification of a gene/s for familial CH and identification of the same genetic variant in related atypical CH cases.
Footnotes
Acknowledgements
The study was supported by grants from the Karolinska Institute and Astra Zeneca. Many thanks to participating families. Thanks to Dr S. Boes-Hansen, Kronobergskliniken in Växjö, to Dr J. Hannerz, formerly at Department of Neurology at Karolinska Hospital in Stockholm, and to Dr M. Linde, Gothenburg Migraine Clinic, for providing us with information about CH families.