
Abstract
The efficacy and tolerability of a CO2-extract of feverfew (MIG-99, 6.25 mg t.i.d.) for migraine prevention were investigated in a randomized, double-blind, placebo-controlled, multicentre, parallel-group study. Patients (N = 170 intention-to-treat; MIG-99, N = 89; placebo, N = 81) suffering from migraine according to International Headache Society criteria were treated for 16 weeks after a 4-week baseline period. The primary endpoint was the average number of migraine attacks per 28 days during the treatment months 2 and 3 compared with baseline. Safety parameters included adverse events, laboratory parameters, vital signs and physical examination. The migraine frequency decreased from 4.76 by 1.9 attacks per month in the MIG-99 group and by 1.3 attacks in the placebo group (P = 0.0456). Logistic regression of responder rates showed an odds ratio of 3.4 in favour of MIG-99 (P = 0.0049). Adverse events possibly related to study medication were 9/107 (8.4%) with MIG-99 and 11/108 (10.2%) with placebo (P = 0.654). MIG-99 is effective and shows a favourable benefit-risk ratio.
Introduction
Feverfew has traditionally been used for the treatment of migraine (1). In recent decades, an increased interest in the ancient herb has developed (2). In the 1980s, two double-blind, randomized, placebo-controlled trials were conducted in patients with migraine. Capsules containing the powdered herb were able to reduce the frequency of migraine attacks (3, 4). The therapeutic effect was significantly superior to placebo. Subsequently, ethanolic extracts of feverfew were tested for efficacy, with contradictory results (5, 6). These trials were difficult to evaluate due to inadequate trial design and low power (7). Ernst concluded that feverfew is likely to be effective in the prevention of migraine. Further clinical studies with feverfew should be performed according to the recommendations by the International Headache Society (IHS) in order to ensure the diagnosis of migraine and a good methodological quality as already mentioned by the IHS in 1988 and in 2000 (8).
The first step in the MIG-99 research project was to develop a stable extract of feverfew. Different methods of feverfew extraction have led to different qualities of extracts regarding stability (9). In contrast to the ethanolic extract, a specially developed feverfew extract using supercritical carbon dioxide (CO2) represents a stable herbal extract highly enriched with parthenolide and thus especially suitable as active ingredient for a pharmaceutical product.
The second step was to investigate the pharmacological effects of feverfew. The mechanism of action of feverfew remains unknown, although several mechanisms have been discussed for its therapeutic effect in migraine (10). Feverfew inhibits the production of prostaglandins, interferes with both contractile and relaxant mechanisms in blood vessels and inhibits the secretion of serotonin (5-HT) from blood platelets (11). These effects may explain the migraine prophylactic properties of feverfew. The mode of action of most drugs used in migraine prophylaxis is not known (12). It is not clear how preventive therapy works, although it seems likely that it modifies the sensitivity of the brain to external and internal stimuli (13).
The next step in this project was to demonstrate the efficacy and tolerability of feverfew in migraine patients. A recent dose finding study showed the efficacy of MIG-99 in reducing the frequency of migraine attacks in migraine at a minimum dose of 6.25 mg extract t.i.d. (14). Despite the statistically significant difference in favour of MIG-99, the results needed to be confirmed and replicated in a second placebo-controlled study in more patients.
Methods
Study design
This phase III study was randomized, multicentre, double-blind and placebo-controlled. A 4-week baseline period without migraine prophylaxis was followed by a 16-week active treatment phase with Tanacetum parthenium CO2-extract (MIG-99) or placebo (Table 1). Study enrolment took place at visit 1 starting period 0 (day − 28; P0) which was a 4-week screening period without prophylactic drug therapy. At the end of this screening phase subjects were randomized (day 0) to one of the two treatment groups, 6.25 mg MIG-99 or placebo t.i.d., and the first period (P1) of the treatment phase started. Thereafter the patients were seen at three control visits every 28 days up to the end of period 4 (day 112; P4). The final visit of the double-blind phase (V6) took place at day 112. Patients had to keep a diary and were instructed to document each migraine attack in terms of date, duration, intensity and any medication taken. Moreover, for each attack they described whether concomitant symptoms, e.g. nausea, vomiting, phono- or photophobia or visual disturbances, had occurred. Furthermore, the patient had to record periods of inability to work, time confined to bed and intake of medication to treat a migraine attack. The completed 4-week diaries were returned to the study centre at each control visit and collected in the case record form (CRF). Patients’ compliance was registered at the study visits 3–6 by counting the returned capsules and blisters. Only patients with an intake between 75% and 125% of the expected number of capsules were eligible for the per-protocol (PP) analysis.
Schedule of investigations and measurements

Inclusion criteria were as follows: male or female out-patients between 18 and 65 years of age with a diagnosis of migraine with or without aura according to IHS criteria (8); migraine attacks for at least 1 year and age at onset < 50 years; an average of three to six migraine attacks per month within the last 3 months prior to study entry; four to six migraine attacks per 28 days within the baseline period; a duration of migraine attacks within the baseline period of 4–72 h; patients’ ability to distinguish between migraine and other headaches; discontinuation of any prophylactic migraine treatment at least 4 weeks (for flunarizine 8 weeks) prior to the beginning of the baseline period.
Exclusion criteria were: hypersensitivity to study medication; pregnancy; intake of analgesics, ergot preparations or triptans for the acute migraine attack on more than 10 days per 4 weeks; > 10 days with headaches other than migraine per month; drug abuse or dependency; expected lack of compliance; psychiatric disorders according to Diagnostic and Statistical Manual of Mental Disorders, 4th Edition (DSM-IV); confirmed diagnosis of gastrointestinal or cardiovascular complaints; other severe diseases including hepatic, renal, gastrointestinal, pulmonary, cardiac, metabolic or endocrine disorders; participation in other clinical trials within the last 3 months or simultaneous participation in another clinical investigation.
The study was performed according to the legal requirements of the German Drug Law, French Drug Law, in accordance with the principles of Good Clinical Practice (GCP) and the latest version of the Declaration of Helsinki. The trial was approved by the ethics committee of the coordinating investigator. Subsequently the ethical review boards of the other centres agreed. The coordinating investigator in France obtained approval of the responsible ethics committee valid for all study centres in France.
Before admission to the clinical study, the nature, scope and possible consequences of the study were explained to the patients by a physician. Only patients who gave written consent after having been informed were included.
Treatment
The patients received either MIG-99 (6.25 mg extract) or placebo three times a day for 16 weeks. The study medications were identical in appearance, size, weight, taste and smell.
The patients were allocated to one of the two treatment groups after the 4-week baseline period by randomization of four in centre-specific blocks on the basis of a randomization code generated by Allphamed, Göttingen, using the validated program Rancode Plus (IDV Datenanalyse und Versuchsplanung, Gauting, Germany). The assignment of random numbers to the patients was carried out in consecutive order according to the time of enrolment into the study, starting with the lowest number of prenumbered medication available within each centre. Patients and investigators remained ‘blind’ throughout the study. All persons active in the study, including the biometrician, remained blinded until the database was locked. Investigators were provided with sealed envelopes containing a code break for emergencies. All envelopes remained sealed throughout the entire study. The intake of medication to treat acute migraine attacks was allowed, but the use had to be documented.
Assessments
At the start of the trial (P0) medical history and migraine history were recorded. A general medical examination was performed. Migraine was classified as migraine with or without aura according to IHS criteria.
The primary efficacy parameter was the total number of migraine attacks within 28 days during the second and third 28-day treatment period (mean of P2 and P3) compared with baseline (prophylaxis-free period P0). This period was selected following the results of the phase II dose finding study. A new migraine attack was assumed if the interval without migraine complaints and headache, which followed the end of the previous attack, lasted at least 24 h. The frequency of migraine attacks was arithmetically adjusted to exactly 28 days by evaluating the last 28 days of the baseline period and by segmenting the treatment phase every 28 days. If the duration of the individual last period was less than 28 days, the interval was replaced by ‘last day documented minus 28 days’. If one or more periods were completely missing, the preceding period was used (last observation carried forward = LOCF).
Predefined secondary efficacy parameters were the number of migraine attacks during each 28-day period of therapy, the clinical global impression of efficacy (very good, good, moderate, none), change of migraine intensity, the total duration of migraine attacks, mean duration of the single attack, number of migraine days (calculated as duration of the whole migraine attack in hours divided by 24 h and rounded up to the next whole number), number of attacks with confinement to bed or inability to work or accompanying migraine symptoms per 28 days, number of migraine attacks with aura per 28 days, type and amount of analgesics and migraine preparations taken and number of drop-outs due to insufficient efficacy.
In addition, the following response criterion was calculated: number of patients with an improvement of > 50% in the number of migraine attacks within 4 weeks.
At the study visits 3–6 vital signs (systolic/diastolic blood pressure and heart rate) and the occurrence of adverse events (AE) were assessed. At visit 6, laboratory parameters (haematology, clinical chemistry, urinalysis) and the clinical global impression of tolerability were assessed. All disturbances of well-being, deterioration of subjective or objective symptoms or signs of known diseases, newly occurred diseases or accidents as well as occurrence of clinically relevant laboratory values outside the reference ranges were regarded as AEs, irrespective of a possible causal relationship to any drugs administered during the trial. AEs which occurred during the baseline phase were also taken into account (Table 1).
Statistics
Primarily, we performed an intention-to-treat (ITT) and additionally a PP analysis. The ITT population was defined as all patients who took study medication after randomization without major violation of inclusion criteria and reporting efficacy at least once. The definite ITT and PP sample was determined after closure of the database and prior to opening the random code. A blind data review was performed to assess which violations of inclusion and exclusion criteria or deviations from the study protocol should be rated as major or minor. Only patients with major protocol violations were excluded from the PP analysis.
An adaptive interim analysis was performed according to Lehmacher and Wassmer (15) using αinterim = 0.0052 and αfinal = 0.0480, preserving an αoverall = 0.05. The final sample size was calculated with the data of the adaptive interim analysis at 72 patients, resulting in 194 patients being enrolled in total. Multivariate linear regression analysis was used for confirmatory statistics. For analysis of the primary endpoint, baseline (P0) was used as covariate and prospectively defined variables as cofactors. Cofactors were photophobia, vomiting, aggravation of headaches by routine physical activity and gender. For sensitivity verification, the efficacy evaluation was repeated by a stepwise backwards multivariate linear regression considering all accompanying symptoms as candidate cofactors (threshold = 0.15). Linear regression analysis, logistic regression analysis (response = reduction in the number of attacks per 28 days > 50%) and other standard methods for comparison of two trial groups were used for the analysis of secondary endpoints (only ITT population). Secondary endpoints were: number of migraine attacks/28 days in the temporal course of the four 28-day intervals of treatment compared with baseline, per cent changes from baseline in the number of attacks/28 days, responders defined as patients with > 50% decrease in migraine frequency, patient and investigator assessment of efficacy.
The study design was jointly developed by the senior authors (H.C.D., V.P.) and the company. The principal investigator had unlimited access to all data.
Results
Patient disposition
The study was performed between April 1999 and April 2002. Ten centres in Germany and four centres in France participated in the trial (see Acknowledgements). A total of 284 patients were included into the baseline period (P0). Two hundred and eighteen patients (MIG-99, N = 108; placebo, N = 110) were enrolled and randomly assigned to one of the two treatment groups. The data of 215 patients could be analysed (MIG-99, N = 107; placebo, N = 108) for drug safety. Forty-five patients were randomized without fulfilling all IHS criteria. This resulted in 170 patients available for efficacy analysis in the ITT sample (89 Tanacetum, 81 placebo). Nine patients were excluded from secondary analysis in the PP set (Fig. 1).

Disposition of patients and datasets (•, radomization).
Demographic characteristics and disease characteristics were similar in both groups (Table 2). The age of the patients ranged from 18 to 64 years with a median of 45.0 years; 82.8% of the patients were female and 17.2% male. Migraine history is shown in Table 3. The median age of first onset of migraine was 20.0 years. The median of migraine attacks per 4 weeks in the last 3 months was five attacks. The most frequent accompanying migraine symptoms were nausea (91.5% of patients), photophobia (92.5%) and phonophobia (82.5%). Neither the per cent proportions nor the averages differed between all patients and the ITT sample. Moreover, any imbalance was controlled by multivariate regression statistics.
Demographic characteristics
Migraine history (mean values; standard deviation)

Demographic data and disease characteristics were not significantly different between the two treatment groups (Tables 2 and 3). Overall, 82.3% of the patients suffered from migraine without aura. The average duration of a migraine attack in the last 3 months was 26 h.
Efficacy
The migraine frequency decreased in the ITT population from 4.8 attacks during baseline to 2.9 (−1.9 ± 0.2) attacks in weeks 5–12 (mean of P2 and P3 adjusted for baseline and the covariables of the primary analysis model) in the verum group and to 3.5 attacks (−1.3 ± 0.2) with placebo. Table 4 shows the results of the statistical analysis for the primary endpoint taking into account the most important covariables. This analysis shows a significantly better efficacy of MIG-99 compared with placebo (P = 0.0456). Table 5 verifies this in another model where only statistically relevant covariables were included. The respective P-values for the analysis of the PP population were P = 0.0771 and P = 0.0467.
Confirmatory statistics of changes in the number of migraine attacks from baseline in the second and third treatment period (intention-to-treat sample; N = 170 for efficacy analyses)

Final model of the stepwise linear regression of changes in the number of migraine attacks from baseline in the second and third treatment period (intention-to-treat)
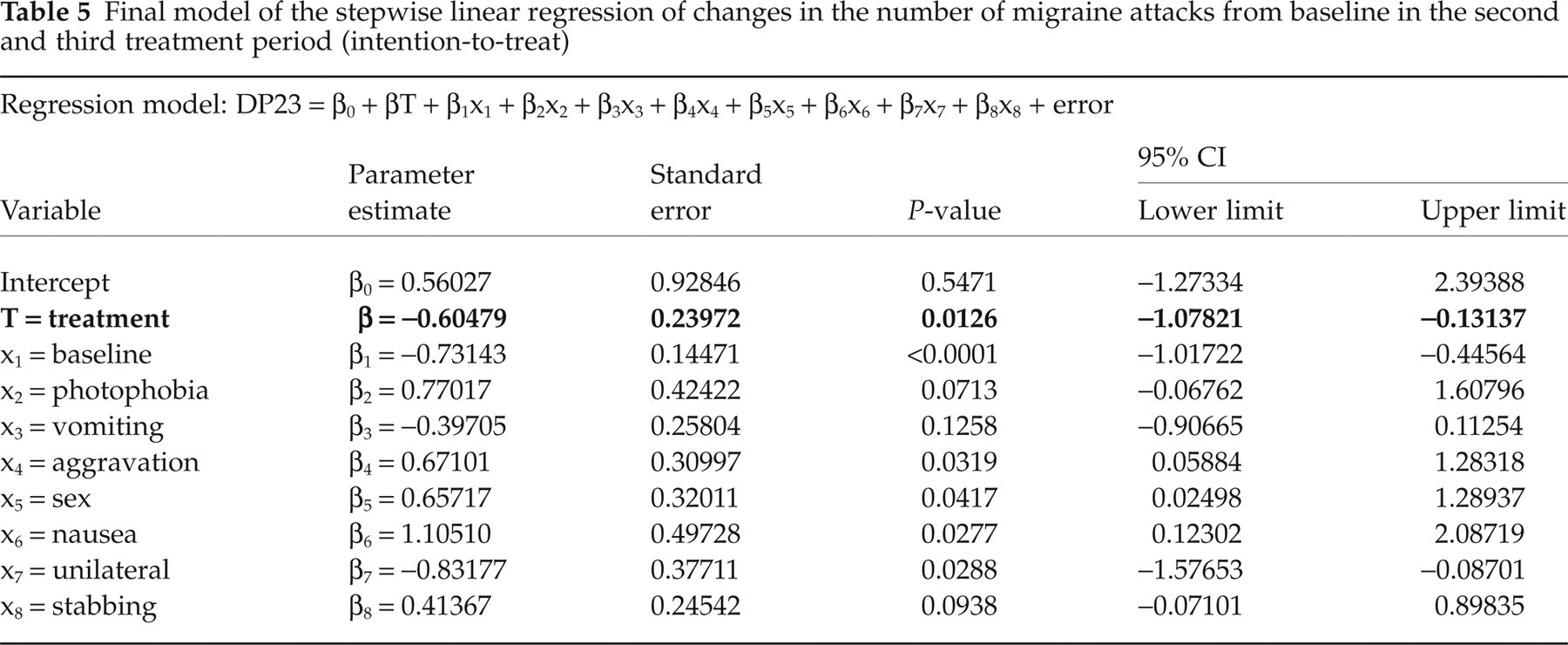
Figure 2 illustrates the temporal course of the number of migraine attacks across the study period adjusted for covariables used in the primary analysis. The adjusted number of migraine attacks was 4.76 attacks at baseline, finally decreasing by 1.9 attacks in periods P1 to P4 in the MIG-99 group, and by 1.3 attacks in the placebo group (P = 0.0148).

Number of attacks in the course of four periods of treatment with MIG-99 (▪, N = 89) and placebo (□, N = 81) (adjusted mean values ± SEM; intention-to-treat sample, last observation carried forward, N = 170).
In the average of periods P2 and P3 the responder rate, i.e. patients with a > 50% decrease of migraine attacks per 28 days compared with baseline, was 30.3% in the MIG-99 group and 17.3% in the placebo group (P = 0.047) (Fig. 3). The response rates adjusted for cofactors represent the data of a ‘typical patient’ in this clinical trial, characterized by: female sex, photophobia, four to five attacks at baseline, nausea, aggravation of headaches by walking stairs or similar routine physical activity, occurrence of headaches in attacks, predominantly unilateral location of headaches, pulsating quality of headaches and with or without stabbing quality of headaches.

Responder rates in the second and third periods of the total intention-to-treat (ITT) sample vs. of the ‘typical patient’ as described by the most frequent combination of cofactors (for details see text). ▪, MIG-99; □, placebo.
The logistic regression of the responder rates, including the same cofactors as identified for the linear analysis, yielded an odds ratio of 3.4 (confidence interval 1.4–7.8; P = 0.0049) in favour of MIG-99.
The median changes from baseline in comparison with the average of period P2 and P3 were analysed for other secondary efficacy parameters. No statistically significant differences between the two treatment groups with respect to nausea, vomiting, photophobia, phonophobia, duration of attacks, accompanying migraine symptoms, visual disturbances, confinement to bed or fitness for work were seen.
The number of migraine days per 28 days decreased in both treatment groups. The reduction in migraine days was higher in the verum group than in the placebo group (Table 6). The group difference in favour of MIG-99 was statistically significant in the repeated measurement test considering the same cofactors as for the primary analysis (P = 0.0353).
Number of migraine days per treatment period (mean and SE).

The total duration of migraine attacks per 28 days decreased from 100 h at baseline to 81, 63, 71 and 65 h in the MIG-99 group and 84, 79, 79 and 84 h in the placebo group in P1, P2, P3 and P4, respectively. The difference was not statistically significant (P = 0.0944).
The global assessment of efficacy revealed a statistically relevant difference between MIG-99 and placebo. Finally, a ‘good’ or ‘very good’ efficacy was assigned by the physicians in 53.8% and 41.4% of patients in the MIG-99 group and placebo group, respectively, and in 53.8% and 38.6% when assessed by the patients. Mantel–Haenszel χ2 test of the temporal course and all four categories of patients’ and investigators’ global assessment resulted in P = 0.024 and P = 0.035, respectively.
Safety and tolerability
Two hundred and fifteen patients were available for the analysis of safety data. Twenty-seven MIG-99-treated (25.2%) and 32 placebo-treated patients (26.6%) experienced AEs in the double-blind treatment phase. Overall, 106 AEs were documented, 56 events in the MIG-99 and 50 events in the placebo treatment group. The rate of patients with AE reported as at least possibly related to study medication was 9/107 (8.4%) following MIG-99 and 11/108 (10.2%) following placebo (P = 0.654). The most frequent AEs were gastrointestinal complaints (MIG-99, N = 4; placebo, N = 7; P = 0.361). These AEs were more frequent in the placebo than in the MIG-99 group. The difference was not significant. The classification by system/organ classes is shown in Table 7. No statistically relevant difference was seen in both medication groups with respect to the profile of events.
Frequency of adverse events
Serious adverse events (SAEs) occurred in five patients in the treatment phase (MIG-99, N = 3; placebo, N = 2). The reason for SAE classification was hospitalization in all cases without any relationship to the study medication.
Overall, six patients discontinued prematurely due to AEs: three in the MIG-99 group and three in the placebo group. During the treatment phase, constipation (N = 2) was the most frequent reason for treatment discontinuation. Abdominal pain was the most frequent reason for treatment discontinuation in the placebo group. The relationship to the study medication was judged as ‘possible’ in one case in each group and as ‘probable’ in two MIG-99 cases and in one placebo case (one case not attributed)
Vital signs showed only slight deviations from baseline without any significant difference between the treatment groups. There were no clinically relevant differences with regard to any laboratory parameter in either treatment group.
Compliance was good: 93.5% of patients in the MIG-99 group and 92.6% of patients in the placebo group were compliant.
The global assessment of tolerability was judged as ‘good’ or ‘very good’ by the physician in 91.5% of patients treated with MIG-99 and in 91.6% in the placebo group. This rate was 92.5% and 92.5%, respectively, when assessed by the patients.
Discussion
The therapeutic efficacy of feverfew has been known for decades. The majority of clinical trials suggested beneficial effects of feverfew compared with placebo (16). Attempts to demonstrate its efficacy have been limited by inadequate trial design and low numbers of patients in the trials (7). This randomized controlled trial demonstrates that a daily oral dose of 6.25 mg MIG-99 t.i.d. is significantly superior to placebo for migraine prophylaxis. These data confirm a recently conducted phase II trial showing that a dosage of 6.25 mg CO2-extract of feverfew t.i.d. is the effective dosage in migraine prevention (14). The effect of MIG-99 begins after 1 month, is maximal after 2 months of treatment and does not decline for at least 4 months. The most pronounced effect was observed with regard to attack frequency, total number of migraine days and migraine duration.
The design of this study was according to the recommendations of the IHS for clinical trials with prophylactic drugs, published in 1991 and updated in 2000 (8). Diagnostic criteria, frequency of attacks, duration of migraine, age of onset, age at entry and gender also corresponded to the recommendations of the IHS. The primary efficacy criterion was defined as the average number of migraine attacks (28 days in period P2 and P3) compared with baseline in a multivariate linear model. We took the mean of periods 2 and 3 because we had data for this period from the phase II trial. Power calculations were based on these earlier results. The choice of the primary efficacy criterion and the kind of evaluation is recommended by the IHS (8). The number of migraine attacks is the single most important and clinically relevant parameter in prophylactic studies (17).
All 170 patients (MIG-99, 89; placebo, 81) fulfilling the inclusion criteria were evaluated in an ITT analysis; 161 patients (MIG-99, 86; placebo, 75) in a valid-case analysis. The number of patients randomized in this study makes it the largest controlled study in migraine prophylaxis with feverfew to be reported to date. This provides a rigorous and robust environment in which to evaluate the effectiveness of feverfew in migraine prophylaxis.
The results showed a statistically significant and clinically relevant difference between MIG-99 and placebo regarding the reduction of migraine frequency. The ITT analysis yielded a decrease in migraine frequency by 1.9 attacks per month by MIG-99 (P = 0.0456 in the primary analysis and P = 0.0126 in the advanced model) when baseline covariables were taken into account. MIG-99 represents an effective prophylactic migraine therapy. The data confirm the results of a recently published clinical trial with MIG-99 (14).
Numerous randomized patients violated the diagnostic inclusion criteria. We were very strict in adhering to the IHS criteria of migraine. We excluded these patients before locking the database. Violators of the inclusion criteria did not differ between groups in the current study.
The effect of placebo in the reduction of frequency and severity of migraine attacks ranged from 11 to 36% in trials investigating migraine prophylaxis (12). The placebo effect in this clinical study was high but within this range. Additional placebo effects might even be desirable in daily practice, provided that the prescribed drug is as safe as possible. Moreover, placebo effects show a marked variability over time, which is also the case in our study (highest mean minus lowest mean = 0.46 attacks per month), whereas the decrease of − 1.9 attacks per month by MIG-99 was nearly constant (highest mean minus lowest mean = 0.09 attacks per month) in months 2–4 (Fig. 2). Moreover, the placebo effect depended on the size of the centre: the more patients per centre, the higher the placebo effect. On the other hand, the efficacy of MIG-99 in the current study was similar to that reported in the first placebo-controlled trial on MIG-99′s efficacy (14).
Patients should take prophylactic migraine agents at least up to 4 weeks before a meaningful clinical response is observed and up to 12 weeks before the maximal effect is realized (18). Therefore, the treatment duration of this clinical trial was 4 months. The first effect of MIG-99 was observed after 1 month and reached its maximum plateau after 2 months of treatment.
The secondary efficacy data confirmed the findings of the primary efficacy parameters. The decrease in migraine days and duration of attacks can be interpreted as an increase of quality of life. In contrast to the total duration of all attacks, the total number of attack days is not flawed by concomitant triptan use (17). Indeed, total number of attack days also showed the significant superiority of MIG-99. Some other secondary parameters were statistically significant, others were not. The trial, however, was not powered to show superiority of MIG-99 in secondary analyses.
The responder rate in this trial was lower than in trials using propranolol (19), divalproex sodium (20) or topiramate (21). Side-effects, however, were less frequent and less severe than in trials using the aforementioned drugs. Therefore the overall benefit/side-effect ratio is still positive. In general, propranolol is recommended as a first-choice preventive medication in migraine therapy (22). A meta-analysis revealed that propranolol caused a 44% reduction in migraine compared with 14% decrease in placebo (19). The data for propranolol show a higher efficacy in contrast to MIG-99. However, it should be considered that baseline migraine frequency was different with the two medications.
Topiramate is a new drug in migraine prevention. Silberstein et al. (23) showed a mean change from baseline in migraine frequency of − 1.5 and − 1.7 under 100 mg/day topiramate in the treatment months 2 and 3. The corresponding data for the placebo group were − 0.6 and − 0.8 migraine attacks. This leads to a group difference of 0.9 attacks on average of months 2 and 3. MIG-99′s reduction in frequency of − 1.9 compared with baseline was achieved under similar baseline frequency. Although the net gain (verum minus placebo) was lower, MIG-99′s efficacy is good and clinically relevant. Topiramate resulted in a much higher rate of drop-outs due to side-effects (21).
The incidence of AEs was comparable in both medication groups. MIG-99 was well tolerated in this study and AEs were mild; similar results were reported by Pfaffenrath et al. (14). Compliance was high (MIG-99, 95%; placebo, 93%). This was probably due to the low number of AEs reported [32 (29.6%) placebo, 27 (25.2%) active medication]. These and all the other safety results demonstrate that MIG-99 provides a safe treatment for migraine prophylaxis. The good tolerability profile contributes to the good compliance rate. Several authors have confirmed that feverfew is safe (24, 16). No major AEs have been documented for feverfew (2). Generally, the incidence of side-effects experienced with feverfew is low. AEs that were reported in clinical studies were generally mild (25). Mouth ulceration and gastrointestinal disturbances are the two main categories of adverse reactions reported to cause discontinuation of use. Mouth ulceration is a rare AE observed in the clinical trials of Johnson et al. (3) and Murphy et al. (4). An explanation of this phenomenon could be that the study medication was powdered with feverfew, so that the patients could develop a contact dermatitis. Inflammation of the mucous membrane of the mouth and tongue, accompanied by swelling of the lips and loss of taste, is probably a contact phenomenon, and is unlikely to be significant with tableted or encapsulated preparations. Apparently this is due to direct contact with the plant and cross-reactions with members of the Compositae rather than sensitization through herbal remedies (26).
The clinical studies with MIG-99 support the statement of the Quality Standards Subcommittee (QQS) of the American Academy of Neurology (AAN), which states that feverfew shows a quality of evidence group 2 (27). In contrast to other feverfew preparations, MIG-99 has a good efficacy and only mild side-effects.
One principle of preventive therapy should be to maximize compliance (22). Therefore, patients’ preferences should be taken into account. Demand for phytotherapeutical therapy has increased in recent decades, especially in Europe. Another factor influencing the compliance rate is tolerability and drug safety. A meta-analysis has shown that one of six patients discontinued propranolol treatment (19). Linde et al. (28) have analysed 58 clinical trials with propranolol and confirmed the high drop-out rate.
In conclusion, this randomized, double-blind, multicentre placebo-controlled, clinical phase III study confirmed the efficacy of the carbon dioxide-based feverfew extract MIG-99 in migraine prophylaxis using 6.25 mg t.i.d. for up to 4 months. The attack frequency was reduced by 1.9 attacks per month compared with baseline. AEs were non-specific and similarly distributed in both treatment groups. This feverfew extract MIG-99 shows a favourable benefit–risk ratio for reducing attack frequency in migraineurs.
Footnotes
Acknowledgements
The following investigators participated in the study: H.-C. Diener and A. Eickermann, Essen; V. Pfaffenrath and M. Singer, München; U. Schutter, Marl; P. F. Donat and M. Pußkeiler, Duisburg; D. Backhaus, Hildesheim; G. Schumann, Bochum; R. Hüntemann, Bochum; J. Peltz, Hattingen; W. Mattern, Bochum; H. Massiou, Paris; M. Jogeix, Bordeaux; J.-M. Visy, Reims; O. Guard and P. Grass, Dijon. The design and conduct of the trial and data analysis were supported by a grant from Schaper & Brümmer, the manufacturer of MIG-99.