
Abstract
Spreading depressions (SD) occur in association with ischaemia, epilepsy and migraine. Intracellular calcium oscillations have been suggested to be involved in the generation and propagation of SD. The present study was performed to study the mechanism of conditioning guinea pig hippocampal slices by the T-type calcium channel blockers NiCl2 and amiloride. SD-like fluctuations of DC potential were recorded by inserting microelectrodes into the CA1 and CA3 regions. The SD occurrence was significantly greater with 10 µmol/l NiCl2 as well as with 25 and 50 µmol/l amiloride than with other concentrations of these substances. The concentration response curve was inversely U-shaped with the maximum repetition rates of SDs being achieved at 10 µmol/l NiCl2 as well as at 25 and 50 µmol/l amiloride. SD occurrence could be completely blocked by the NMDA antagonist APV (10 µmol/l) in all cases. These data demonstrate that modulation of the Ca2+ dynamics conditioned guinea pig hippocampal slices and increased their susceptibility to generate SD.
Introduction
Spreading depression (SD), as initially described by Leão (1), is characterized by a self-propagating front of depolarization, associated with a depression of the neuronal bioelectrical activity. This phenomenon has been studied in vivo in several animal species and in vitro in brain slices and retinal preparations under various experimental conditions. There are reasons to believe that SD is involved in some clinical disorders, including migraine, head injury and cerebral ischaemia (2, 3).
Local propagating translocations of ions are commonly agreed to underlie this potential change. There is a massive release of K+ from the cells into the extracellular fluid and an entry of Cl−, Na+ and Ca2+ into the cells (4). It was shown that SD is associated with calcium dynamics (5, 6). It represents an extracellular decrease and intracellular increase of calcium ions immediately after the onset of the transient membrane depolarization (7). Consequently a transmembranous calcium ion flux was suggested to play a role in SD initiation and propagation (8). In fact, manipulation of the calcium composition such as local application of calcium chelating agents like EDTA induced SD (9). Furthermore, some investigations pointed out that the blocker of L-type calcium channels, verapamil, induced SD in rat hippocampal slices (10). It was suggested that increased uptake of 3H-nimodipine during transient depolarization of SD could be an indication of calcium channel modulation (11). In line with the aforementioned findings is the suggestion that calcium channels are involved in the pathophysiological processes of migraine (12, 13). Pharmacological analysis in guinea pig hippocampal tissues has identified N-, L-, and putative P-type Ca2+ channels in addition to a T-type channel (14, 15). Low concentrations of NiCl2 (< 100 µmol/l) and amiloride (< 500 µmol/l) are supposed to act as T-type calcium channel blockers (14, 16, 17). A recent study has shown that application of low concentrations NiCl2 and amiloride elicited repetitive SD in human neocortical tissues (18). To address the mechanism of neural tissue conditioning by T-type calcium channel blockers to induce SD, the present study set out to investigate SD elicited by NiCl2 and amiloride in guinea pig hippocampal slices.
Materials and methods
The experiments were carried out on hippocampal slices from guinea pig (300–400 g). The brain was removed under deep methohexital anaesthesia. The hippocampi were dissected and cut into slices of 500 µm thickness. The slices were preincubated at 28°C for 60 min in artificial cerebrospinal fluid (ACSF). The ACSF contained (mmol/l): NaCl 124, KCl 4, CaCl2 1.0, NaH2PO4 1.24, MgSO4 1.3, NaHCO3 26 and glucose 10. The ACSF was continuously equilibrated by 5% CO2 in O2 and the pH stabilized at 7.35–7.4. After 30 min preincubation, CaCl2 was elevated to 2.0 mmol/l. The slices were transferred to an interphase recording chamber and perfused with ACSF at 32°C. Field potentials were recorded with glass microelectrodes (2 mol/l NaCl; 2–10 MΩ) from stratum pyramidale in CA1 and CA3 regions. The reference electrode and the connection to the microelectrode were symmetric Ag–Ag–KCl bridges. Field potentials were traced by an inkwriter and stored by a digital oscilloscope. The Schaffer collaterals were stimulated by a bipolar platinum electrode (trains of six pulses; interstimulus interval 1 min; applied every 30 min). Stimulus intensity was adjusted to half the intensity needed to elicit maximum population spike amplitude in CA1.
Two experimental protocols were used, each of which consisted of several periods. First experimental protocol: (Period 1) control 1; superfusion with ACSF (30 min), test for spontaneous epileptiform discharges and for spontaneously appearing SD; (Period 2) superfusion of amiloride (25–200 µmol/l; 5 h) or NiCl2 (1–50 µmol/l; 5 h) with and without amino phosphonovaleric acid (APV; 10 µmol/l); (Period 3) control 2. Second experimental protocol: (Period 1) control 1; (Period 2) pressure injection of KCl solution (3 mol/l) through microelectrodes into slices (electrode tip diameter 2 µm, 0.5–2.0 bar; 200–800 ms; up to 2 separate injections); (Period 3) same procedure as in (Period 2) after application of APV (10 µmol/l; 60 min); (Period 4) wash out of APV by superfusion of ACSF (30 min); (Period 5) superfusion of amiloride (50 µmol/l) or NiCl2 (10 µmol/l; 5 h).
SD-like events were evaluated with respect to their amplitudes, duration and repetition rates (number of SD-like waves/5 h). All values given represent mean ±
Results
Each experiment started with 30 min superfusion of the slices with control ACSF (protocols 1 and 2; Period 1) to detect spontaneously occurring epileptiform field potentials (EFP) or SD-like fluctuations. In all slices there were none of either (n = 72).
Addition of NiCl2 in concentrations of 1, 2.5, 5, 10, 25 and 50 µmol/l (n = 36; six for each concentration) to control ACSF (protocol 1, Period 2) elicited negative fluctuations of DC potential which were sometimes followed by positive shifts (Fig. 1a1). The same DC potential changes appeared when amiloride in concentrations of 25, 50, 100 and 200 µmol/l (n = 24; six for each concentration) was given to control ACSF (protocol 1, Period 2; Fig. 1b1). Negative deflection and successive positive shifts usually appeared in CA1 and CA3 regions with a time lag ranging from 5 s to 30 s (Fig. 2). Application of APV (10 µmol/l; protocol 1, Period 2) together with NiCl2 (10 µmol/l) or amiloride (50 µmol/l) prevented any SD-like fluctuations (n = 6; Fig. 1a2, b2). After washout of NiCl2 or amiloride, SD failed to occur (protocol 1, Period 3). DC fluctuations elicited by superfusion of NiCl2 and amiloride were of different shapes in different experiments (Fig. 3). This was predominantly due to the variable expression of the waves following the early stereotypical negative deflections. The shapes of the deflections at the two recording sites were always very similar.

Typical spreading depression (SD)-like fluctuations of DC potential during superfusion with NiCl2 and amiloride and failing of appearance with amino phosphonovaleric acid (APV) in guinea pig hippocampal slices. (a1–b2) Different slices. (a1,b1) Appearance of SD-like fluctuations with NiCl2 (10 µmol/l;a1) and amiloride (50 µmol/l;b1). (a2,b2) SD-like fluctuations failed to appear when NiCl2 (a2) and amiloride (b2) were simultaneously applied with APV (10 µmol/l). The points of time given refer to commencements of substance application.

Suppression of evoked potentials (EP) during spreading depression (SD)-like fluctuations of DC potential in a guinea pig hippocampal slice. DC potentials were recorded from CA1 and CA3 regions. Induction of SD-like wave by superfusion with amiloride (25 µmol/l). EP were elicited by single electrical stimuli applied to Schaffer collaterals. Point of time given refers to commencement of amiloride application. EP1 and EP2 directly related by arrows to the DC recording; EP3, 30 min after SD. Time lag between SD appearance in CA1 and CA3 regions is indicated by vertical broken lines.
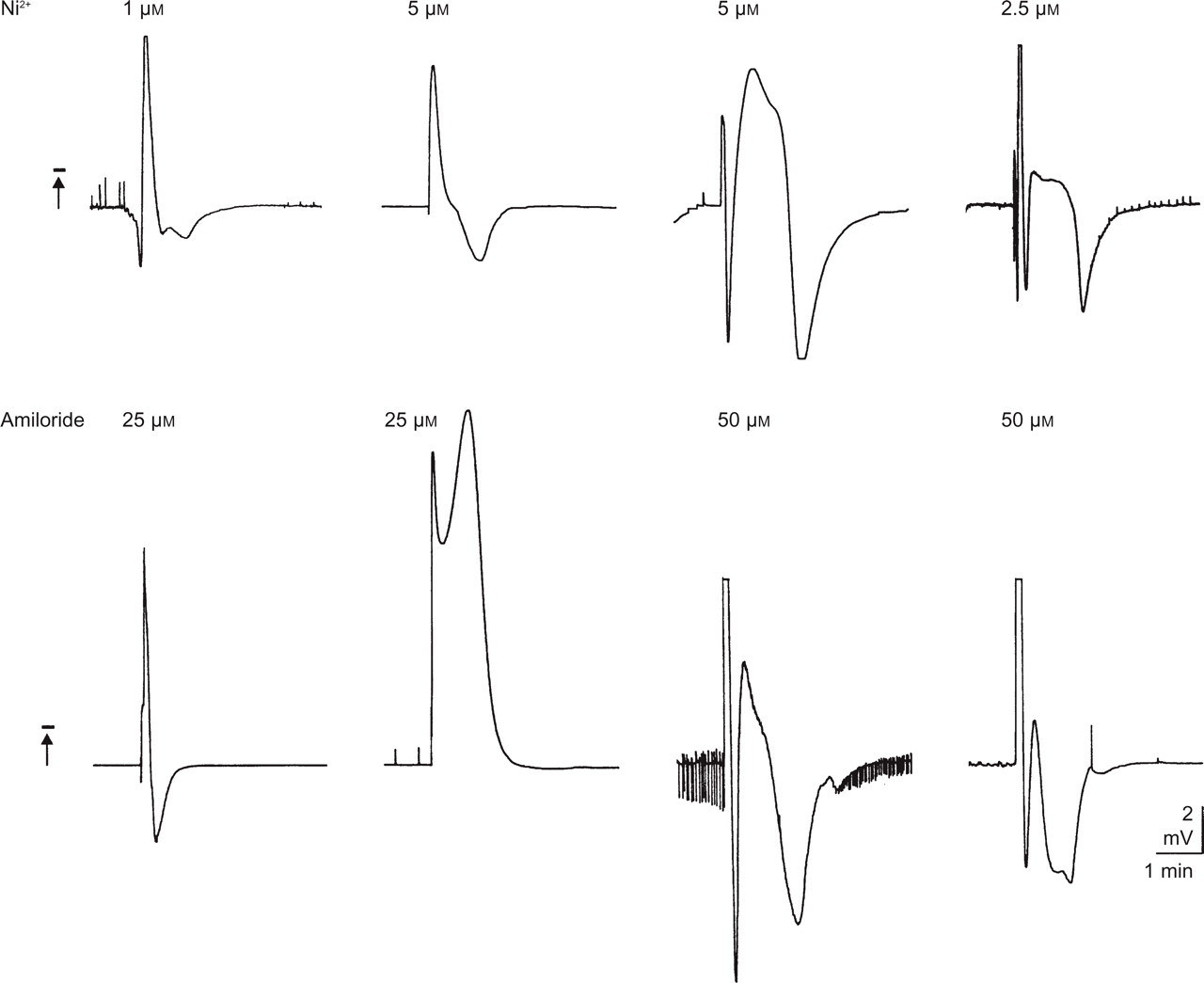
Differential shapes of spreading depression (SD)-like fluctuations of DC potential in guinea pig hippocampal slices. Induction of SD-like waves by superfusion of NiCl2 and amiloride in different concentrations.
Amplitudes, durations and repetition rates of the aforementioned SD-like DC potential shifts are shown in Table 1. Amplitudes and durations did not differ significantly with the concentrations of NiCl2 and amiloride. The repetition rates of SD-like DC fluctuations, however, were significantly greater with 10 µmol/l NiCl2 as well as with 25 and 50 µmol/l amiloride than with other concentrations of these substances (Fig. 4; P = 0.001–0.002; Table 1).
Properties of spreading depression (SD)-like fluctuations of DC potential induced in guinea pig hippocampal slices with superfusion of NiCl2 and amiloride
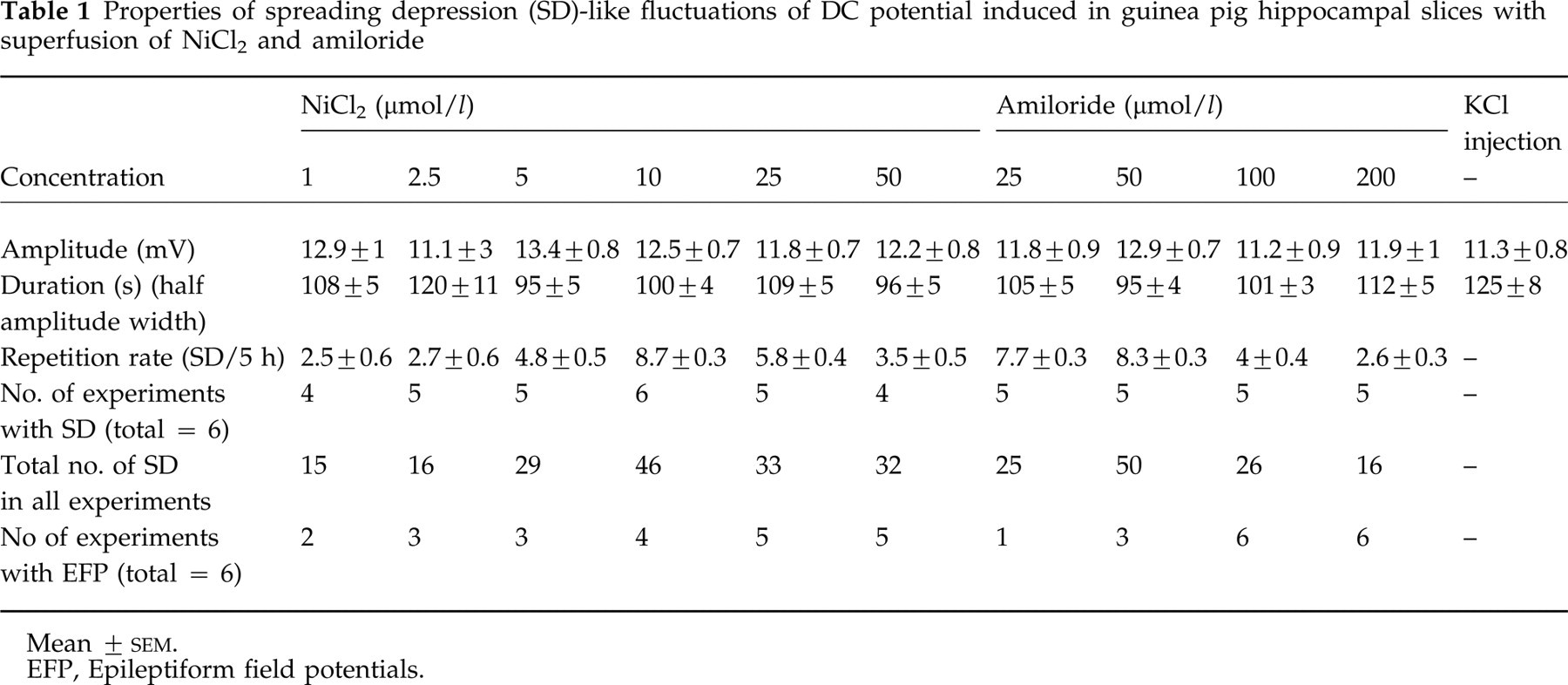
Mean ±
EFP, Epileptiform field potentials.
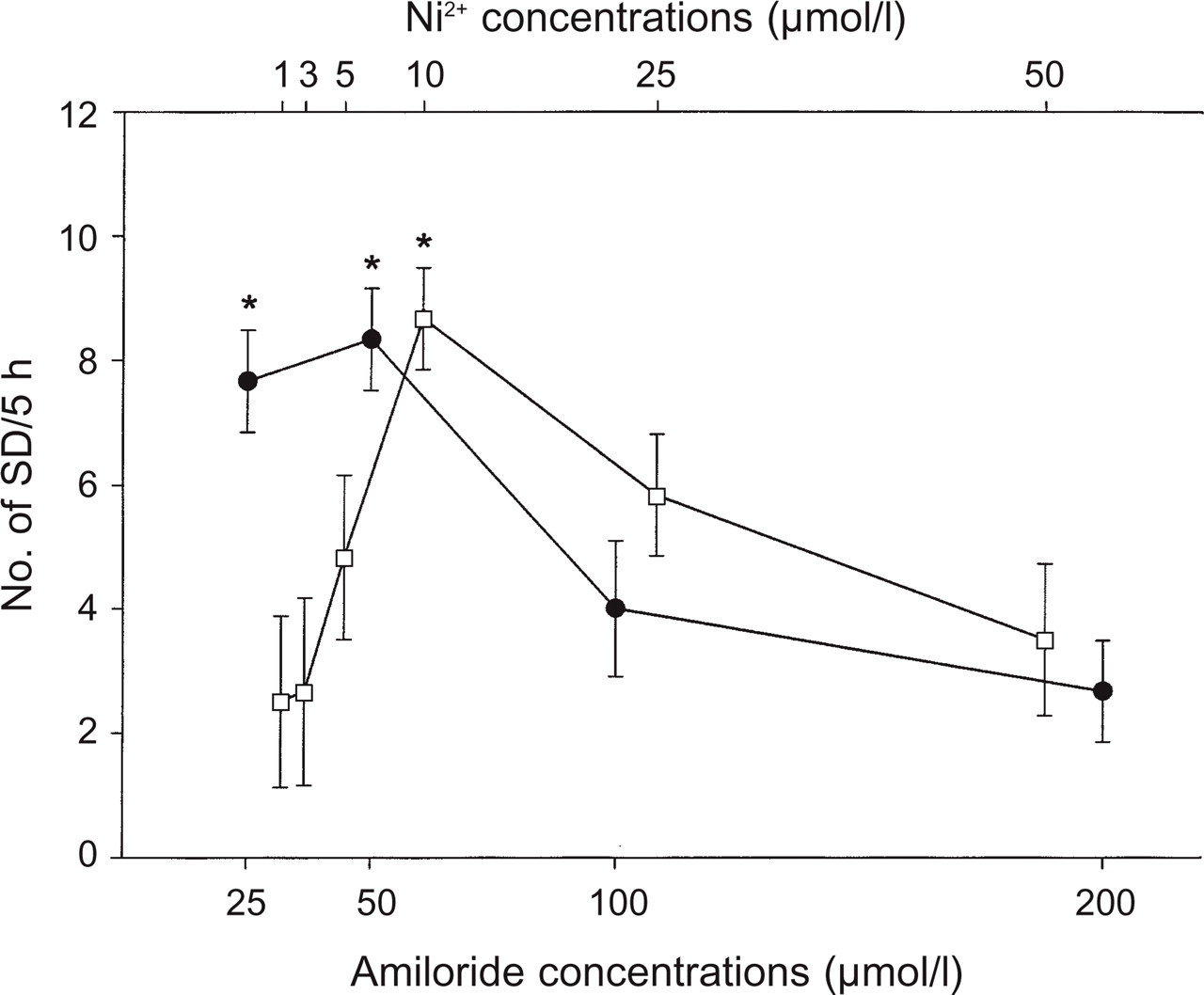
Dose dependency of the number of spreading depression (SD)-like negative DC potentials in the presence of NiCl2 (□) and amiloride (●). The repetition rates of SD-like fluctuations with 10 µmol/l NiCl2 as well as with 25 and 50 µmol/l amiloride application are significantly greater than with other concentrations (∗significant at P < 0.05; mean ±
During superfusion with NiCl2 and amiloride, some slices showed spontaneous EFP (Table 1). They started after 30 min and disappeared after another 3–4 h. There was no relation between the appearance of EFP and SD-like DC potential changes. The EFP were either found to commence before SD-like DC shifts appeared for the first time, or they ran in parallel with DC changes or already ceased before the first DC potential waves appeared. In the case of synchronized appearances of both phenomena, EFP were suppressed during SD-like fluctuations.
Evoked potentials elicited by Schaffer collateral stimulation and recorded in CA1 region were transiently depressed during appearance of SD in all cases. They reappeared a few minutes after commencement of SD-like waves and recovered completely after about 30 min (Fig. 2).
Injection of KCl (3 mol/l) into hippocampal slices (protocol 2, Period 2; n = 6) elicited SD-like fluctuations of the DC potential. They represented initial negative shifts which were followed by positive ones (Fig. 5a1, b1). The amplitude and duration of these SD-like fluctuations are given in Table 1. There was no difference in the SD-like waves elicited by NiCl2 and amiloride. Application of APV (10 µmol/l; protocol 2, Period 3) prevented KCl-induced negative fluctuations (Fig. 5a2, b2). In these slices application of NiCl2 (10 µmol/l; protocol 2, Period 5) or amiloride (50 µmol/l) induced SD-like fluctuations which were similar in shape to those evoked by KCl injection (Fig. 5a3, b3).

Appearance of spreading depression (SD)-like fluctuations of DC potentials after injection of KCl and after superfusion with NiCl2 and amiloride in guinea pig hippocampal slices. (a,b) Two different slices. (a1,b1) Injection of KCl solution (3 mol/l) via a microelectrode-elicited SD-like DC fluctuation during superfusion with artificial cerebrospinal fluid (ASCF). (a2,b2) Superfusion of amino phosphonovaleric acid (APV; 10 µmol/l) prevented the induction of SD-like fluctuations by KCl injection. (a3,b3) In the same slices, superfusion with NiCl2 (10 µmol/l) and amiloride (50 µmol/l) induced SD-like waves of similar shape as with KCl injection. The points of time given refer to commencement of substance application.
Discussion
The present results indicate that NiCl2 and amiloride are able to condition guinea pig hippocampal slices to express spontaneous SD-like fluctuations of DC potential. The repetitive spontaneous SD were observed after long-term application of the compounds mentioned above. Application of the NMDA antagonist APV prevented all SD. It was shown that long-term application of NiCl2 (10 µmol/l) and amiloride (50 µmol/l) can elicit cortical SD in human neocortical tissues (18). The DC potential shifts in question can be assumed to reflect a classical SD phenomenon for the following reasons: (i) DC potential shifts are spreading since there is a time lag between the onset of SD-like fluctuations in different sites within a slice (1). The velocity of SD propagation, however, can not reliably be measured in the present case since SD had been elicited by superfusing the whole slice with NiCl2 and amiloride which leaves the site of origin of SD unidentified; (ii) the DC shifts did not occur in the presence of NMDA antagonist APV which is commonly agreed to suppress SD (19–21); (iii) evoked potentials were transiently reduced in amplitude with DC fluctuations and recovered within minutes (1); (iv) the shape of the spontaneous DC waves elicited by NiCl2 and amiloride resembled precisely those induced by injection of KCl which represent the classical trigger of SD.
Low concentrations of NiCl2 (< 100 µmol/l) and amiloride (< 500 µmol/l) are supposed to act as T-type calcium channel blockers (14, 16, 17). With higher concentrations, however, both NiCl2 and amiloride are supposed to exert additional effects. Thus, some studies have shown that NiCl2 at higher concentrations (≥ 300 µmol/l) blocks the L-type calcium channels and amiloride at higher concentrations (> 500 µmol/l) inhibits high threshold Ca2+ current and Na + –K + exchanger (16). In guinea pig hippocampal tissue, the number of SD in the presence of NiCl2 and amiloride showed a clear dose-dependency of an inversed U-shape (Fig. 4). The lowest concentrations of the substances to elicit SD in the present study (NiCl2, 1 µmol/l; amiloride, 25 µmol/l) and the concentrations with the highest rate of appearance of SD (NiCl2, 10 µmol/l; amiloride, 25 and 50 µmol/l) are in the concentration range of NiCl2 and amiloride in which T-type calcium channel blockage has been demonstrated. Therefore, it can be assumed that the modulation of the Ca2+ dynamics by NiCl2 and amiloride conditioned guinea pig hippocampal slices and increased their susceptibility to generate SD.
NiCl2 and amiloride application has conditioned the tissue and enhanced its susceptibility to generate SD. Two different mechanisms might be taken into consideration: (i) subsequent to the NiCl2 and amiloride treatment a slow but constant decrease of intracellular calcium concentration may have led to a calcium-mediated enhancement of the NMDA response (22) and thus to the initiation of SD via the glutamatergic pathway (23); (ii) eliciting of SD by some calcium channel blockers, like NiCl2, amiloride or verapamil, may be due to a blockade of transient potassium current (A current). It is well known that inorganic and organic calcium channel blockers in different neurones reduce the amplitude of A current (24–26). This effect is found to lead to an accumulation of K + in the extracellular space on the basis of an increased GABA release (27).
Data demonstrated that although elevation of intracellular and reduction of extracellular calcium are associated with SD (6, 7), such calcium dynamics are not critical to induction and propagation of that in slice preparations (28, 29). However, it should be noted that in the retina, SD is inhibited in the absence of external calcium or by a non-specific calcium channel blocker (30, 31). SD has some clinical interest since they have been suggested to occur during the aura phase of the migraine attacks (32). Such attacks have been found to be counteracted by prophylactic treatment with calcium channel blockers (33). It was suggested that these substances enhance the threshold for eliciting of SD (34). The optimum effect needed often a few months of treatment (35). On the other hand, with commencement of treatment one of the most common acute side-effects caused by application of calcium channel blockers is headache. In this context, it should be noted that, besides amiloride and NiCl2, L-type calcium channel blockers, e.g. verapamil, also can induce SD (10). Thus, it may be suggested that calcium dynamics play a dual or even a paradoxical role in eliciting as well as in preventing SD.
There is evidence that neural tissue has the ability to stop SD at their very beginning. The mechanism by which this ability is lost has not yet been clarified (36). In respect to this, it may be of interest that studies have shown that initiation of SD is more difficult in human than rodent cortex (37). Thus, a higher number of SD is generated in the guinea pig in the presence of NiCl2 and amiloride than in man (18). This may be due to properties of the tissue as far as the general cell density or receptor distribution is concerned (38).