
Abstract
Glutamate uptake is reduced during ischemia because of perturbations of ionic gradients across neuronal and glial membranes. Using immunohistochemical and Western blot analyses, the authors examined the expression of the glutamate transporters EAAC1, GLAST, and GLT-1 in the rat hippocampus and cerebral cortex 8 hours and 1 to 28 days after transient forebrain ischemia. Densitometric analysis of immunoblots of CA1 homogenates showed a moderate increase in EAAC1 protein levels early after the insult. Consistently, it was observed that EAAC1 immunostaining in CA1 pyramidal neurons was more intense after 8 hours and 1 day of reperfusion and reduced at later postischemia stages. A similar transient increase of EAAC1 immunolabeling was detected in layer V pyramidal neurons of the cerebral cortex. In addition, the authors observed that EAAC1 also was located in oligodendroglial progenitor cells in subcortical white matter. The number of EAAC1-labeled cells in this region was increased after 3 and 28 days of reperfusion. Finally, changes in GLAST and GLT-1 expression were not observed in the CA1 region after ischemia using immunohistochemical study or immunoblotting. Enhanced expression of EAAC1 may be an adaptive response to increased levels of extracellular glutamate during ischemia.
Glutamate is the most widely distributed excitatory amino acid in the mammalian brain, and its uptake is essential to prevent excitotoxic death. Altered glutamatergic neurotransmission has been implicated in numerous biological disorders, since an excessive activation of glutamate receptors has been demonstrated to lead to neuronal death (reviewed in Obrenovitch and Urenjak, 1997). The concentration of glutamate at the synaptic cleft is maintained by high-affinity, sodium-dependent glutamate transporters located in the plasma membranes of neurons and astrocytes surrounding synapses. At least five different glutamate transporters have been cloned. The GLAST and GLT-1 glutamate transporters are located principally in glial cells; EAAC1 and EAAT4 are expressed in neurons, and EAAT5 transporters are confined to the retina (reviewed in Kanai et al., 1997). GLT-1 appears to be a major determinant of glutamate homeostasis, since inhibition of its synthesis by specific antisense oligodeoxynucleotides results in neuronal excitotoxic cell death (Rothstein et al., 1996). Likewise, knockout mice lacking GLT-1 show lethal spontaneous seizures and increased susceptibility to acute cortical injury (Tanaka et al., 1997). In contrast, EAAC1-knockout mice develop dicarboxylic amino aciduria and behavioral abnormalities but show no sign of neurodegeneration (Peghini et al., 1997).
Transient ischemia causes molecular alterations, which may contribute to neuronal hyperexcitability and cell death, particularly in vulnerable regions of the brain such as the hippocampal CA1 area (Choi, 1996; Luhmann, 1996). Ischemic insults are characterized by a failure of energy supply to the cells and, as a consequence, a rundown in transmembrane electrochemical gradients. Because the transmembrane gradient of co-transported ions constitutes the driving force of glutamate transporters, its alteration may result in a transient reversal of the transport (Attwell et al., 1993; Szatkowski and Attwell, 1994). Thus, ischemia leads to an increased concentration of extracellular glutamate (over 100 μmol/L; Hagberg et al., 1985), which can cause excitotoxic neuronal death before the extracellular glutamate concentration returns to near-normal levels on recirculation (Silver and Erecinska, 1992). Other mechanisms involved in ischemic damage may include potentiation of N-methyl-D-aspartate receptor-mediated currents, a disturbance of calcium homeostasis, and an increase in oxygen free radical formation (Choi and Rothman, 1990).
The expression of glutamate transporters is differentially regulated. Thus, neuronal soluble factors induce the expression of GLT-1 (Swanson et al., 1997), and glutamate receptor agonists upregulate GLAST transporter expression in astrocytes (Gegelashvili et al., 1996). Furthermore, disruption of glutamatergic pathways leads to down-regulation of GLAST and GLT-1 expression but not to that of EAAC1 (Ginsberg et al., 1995; Levy et al., 1995). Taken together, these data raise the possibility that the changes occurring in ischemia may have profound effects on glutamate transporter expression, which can contribute to exacerbating or improving the lesions induced by the insult. Therefore, understanding the regulation of glutamate transporter expression after ischemia is critical. To that end, we analyzed by Western blot and immunohistochemical analyses the expression of the glutamate transporters EAAC1, GLAST, and GLT-1 in the hippocampus, cerebral cortex, and in periventricular white matter at several stages of reperfusion after transient forebrain ischemia.
MATERIALS AND METHODS
Surgery
Transient forebrain ischemia was induced by the four-vessel occlusion method (Pulsinelli and Brierley, 1979) in male Wistar rats (body weight 250 to 300 g). The animals were anesthetized with pentobarbital (25 mg/kg intraperitoneally), and the vertebral arteries were electrocauterized in the alar foramina of the first cervical vertebra. Immediately after this procedure, an incision was made to isolate the common carotid arteries, and polyethylene cuffs were placed loosely around each artery without interrupting blood flow. The incision then was closed with surgical clips, and the animals were allowed to recover from the anesthesia overnight. The next day, rats were lightly reanesthetized with ether and the surgical clips removed. When the level of anesthesia was such that the animals did not show spontaneous movement but reacted to pain stimuli, the common carotid arteries were clamped for 20 minutes. Criteria for forebrain ischemia were bilateral loss of the righting reflex, paw extension, and mydriasis. Animals who did not fully loose their righting reflexes or who developed seizures after carotid artery occlusion were excluded from the study. All efforts were made to minimize animal distress and to reduce the number of animals used.
Immunohistochemistry
Expression of glutamate transporters was examined by immunohistochemistry (n = 3 to 5 animals per experimental group) using high-affinity purified polyclonal rabbit antibodies to C-terminal EAAC1, N-terminal GLAST, and C-terminal GLT-1 epitopes, generously provided by Dr. J. D. Rothstein (Rothstein et al., 1994, 1996). Tissue fixation, sectioning, and processing for conventional immunoperoxidase histochemical study were carried out as previously described (Gottlieb and Matute, 1997). A preliminary evaluation of postischemic damage was carried out in each brain using toluidine blue-counterstained coronal sections containing the dorsal hippocampus. Antibodies were used at the following concentrations: C-EAAC1, 66 ng/mL; N-GLAST, 40 ng/mL; and C-GLT-1, 37 ng/mL. The primary antibodies were detected using biotinylated goat anti-rabbit IgG (1:200; Vector Laboratories, Burlingame, CA, U.S.A.) and the Vectastin ABC-Elite Kit (Vector) with 3,3′-diaminobenzidine tetrahydrochloride as a peroxidase substrate. Negative controls in all experiments included the omission of the primary antibody and incubation with control and nonimmune IgG, and yielded no labeling. The results obtained with commercially available antibodies to EAAC1 and GLAST (both diluted 1:2,000; Chemicon, Temecula, CA, U.S.A.) were similar to those found with the corresponding antisera provided by Dr. Rothstein.
Double-immunofluorescent labeling experiments were carried out sequentially with antibodies to the glutamate transporter EAAC1 (0.6 μg/mL) and to glutathione-S-transferase Yp subtype (GST-Yp; 1:500; Biotrin Int., Ireland) or the NG-2 chondroitin sulfate proteoglycan (0.8 μg/mL; Levine et al., 1993). After blocking nonspecific binding with 4% normal goat serum, 0.1% Triton X-100, and 0.02% sodium azide in phosphate-buffered saline, the sections were incubated with antibodies to EAAC1 overnight at 4°C. The primary antibody was viewed with a fluorescein isothiocyanate-conjugated goat antirabbit IgG (Alexa 4088; 1:200; Molecular Probes, Eugene, Oregon, U.S.A.). Next, the sections were incubated first with 8% normal goat serum, as described earlier, and then with antibodies to GST-Yp or to NG-2 for 1 hour at 37°C. Finally, they were incubated at room temperature for 1 hour with biotinylated antibodies to rabbit IgG (1:200; Vector) followed by a streptavidin-tetramethyl rhodamine isothiocyanate-conjugate (1:100; Chemicon). Negative controls in all experiments included the omission of one of the primary antibodies and yielded no immunofluorescence with the corresponding detection procedure, indicating the reliability of the protocol assayed, as previously described (Domercq and Matute, 1999).
Cell counting
We quantified the number of EAAC1+ cells in periventricular subcortical white matter in control animals and in animals subjected to transient forebrain ischemia after 1 to 28 days of reperfusion (n = 3 animals per experimental group). Counts were taken from the right and left hemisphere in each immunostained section, two sections for each experiment, and three independent experiments. Labeled cells were counted by two different observers blinded with respect to the experimental groups. The number of positive cells was expressed as cells per square millimeter. The surface area of each counted field was calculated using the National Institutes of Health Image program on the drawings taken by camera lucida of the region studied. All the tissue sections analyzed were 30 μm in depth and were obtained using the same vibratome. Statistical comparisons were performed by repeated-measures analysis of variance with a Fisher's protected least significance difference post hoc analysis. Data are represented as the mean ± SEM of n = 3 independent experiments.
Western blot analysis
Rats were deeply anesthetized with chloral hydrate and decapitated 8 hours or 1, 3, 7, and 28 days after reperfusion. Nonoperated animals were used as controls. Hippocampi were removed from the brain at 4°C, and the CA1 region then was dissected out immediately. Tissue was homogenized in 20 mmol/L Tris-HCl (pH 7.4), 10% sucrose, 1 mmol/L ethylenediamine tetraacetic acid, 5 mmol/L EGTA, 1 mmol/L phenylmethylsulfonic fluoride, 10 mmol/L benzamidine, 20 μg/mL leupeptin, and 0.1 μmol/L pepstatin with a Dounce homogenizer (Heidolph, Germany). After centrifugation of the homogenate (10,000 g, 30 minutes), the supernatants were diluted 1:1 with an electrophoresis sample buffer (Laemmli, 1970) and then boiled for 5 minutes. Proteins were separated by SDS-polyacrylamide (7.5%) gel electrophoresis and subsequently transferred to polyvinylidene fluoride membranes using a semidry electroblotter (Sartorius, Germany). Nonspecific binding to the membrane was blocked for 1 hour at room temperature with 10% nonfat milk and 2% bovine serum albumin diluted in 50 mmol/L Tris-HCl, 200 mmol/L NaCl, and 0.1% Tween-20 pH 7.4. Blots were incubated for 2 hours at room temperature or overnight at 4°C with affinity purified antibodies to EAAC1 (0.6 μg/mL), GLAST (0.2 μg/mL), and GLT-1 (0.034 μg/mL) in the same buffer containing 1% nonfat milk. After washing, blots were finally incubated with horseradish-peroxidase conjugated anti-rabbit antibody (1:1000, Sigma, St. Louis, MO, U.S.A.). Immunoreactive proteins were visualized with an enhanced chemiluminescence substrate (Super Signal ULTRA, Pierce, Rockford, IL, U.S.A.). Densitometry using the NIH Image program of the blots was used to quantify the temporal changes in transporter levels. For each antibody, standard curves were generated using increasing concentrations of CA1 protein homogenates. Statistical analysis was carried out as described in the previous section.
RESULTS
The histopathologic methods of the postischemic brains used in this study were comparable to those described earlier (Pulsinelli et al., 1982). Thus, damage to the hippocampus was not obvious at postlesion times of 8 hours and 1 day, respectively. However, most neurons in the CA1 pyramidal cell layer were severely impaired 3 days after the ischemic insult and virtually disappeared at longer recirculation periods, which is consistent with findings reported under the experimental conditions used in the current study (refer to Gottlieb and Matute, 1997, 1999).
Altered expression of the glutamate transporter EAAC1 after ischemia
To examine the possible changes in glutamate transporter expression from ischemic-induced lesions, we conducted immunohistochemical study with antisera to the EAAC1, GLAST, and GLT-1 transporters in the hippocampus after 8 hours and 1 to 28 days after reperfusion. The distribution of these three transporters in control animals was in agreement with previous immunohistochemical studies (Rothstein et al., 1994; Lehre et al., 1995). Therefore, the results of the current study in control hippocampus are described briefly and are used as a reference for comparison with those observed after transient ischemia.
In control brains, EAAC1 was present in the hippocampal neuropil, and its expression was selectively enriched in pyramidal neurons (Figs. 1A and 1B). At 8 hours and 1 day after ischemia, we observed an increase of EAAC1 immunoreactivity in cell bodies and the proximal part of apical dendrites in CA1 pyramidal neurons (Fig. 1C to Fig. 1F). However, EAAC1 immunostaining in CA1 pyramidal neurons was drastically reduced at 3 days after ischemia and thereafter as a result of the neuronal death caused by the insult (Fig. 1G to Fig. 1I). A weak increase in EAAC1 expression also was observed in other hippocampal regions after ischemia. This was particularly obvious in pyramidal cells and in the stratum oriens of CA3 at 1 day of recirculation (Fig. 1E), and EAAC1 levels returned to control values at 3 postlesion days and thereafter (not shown).
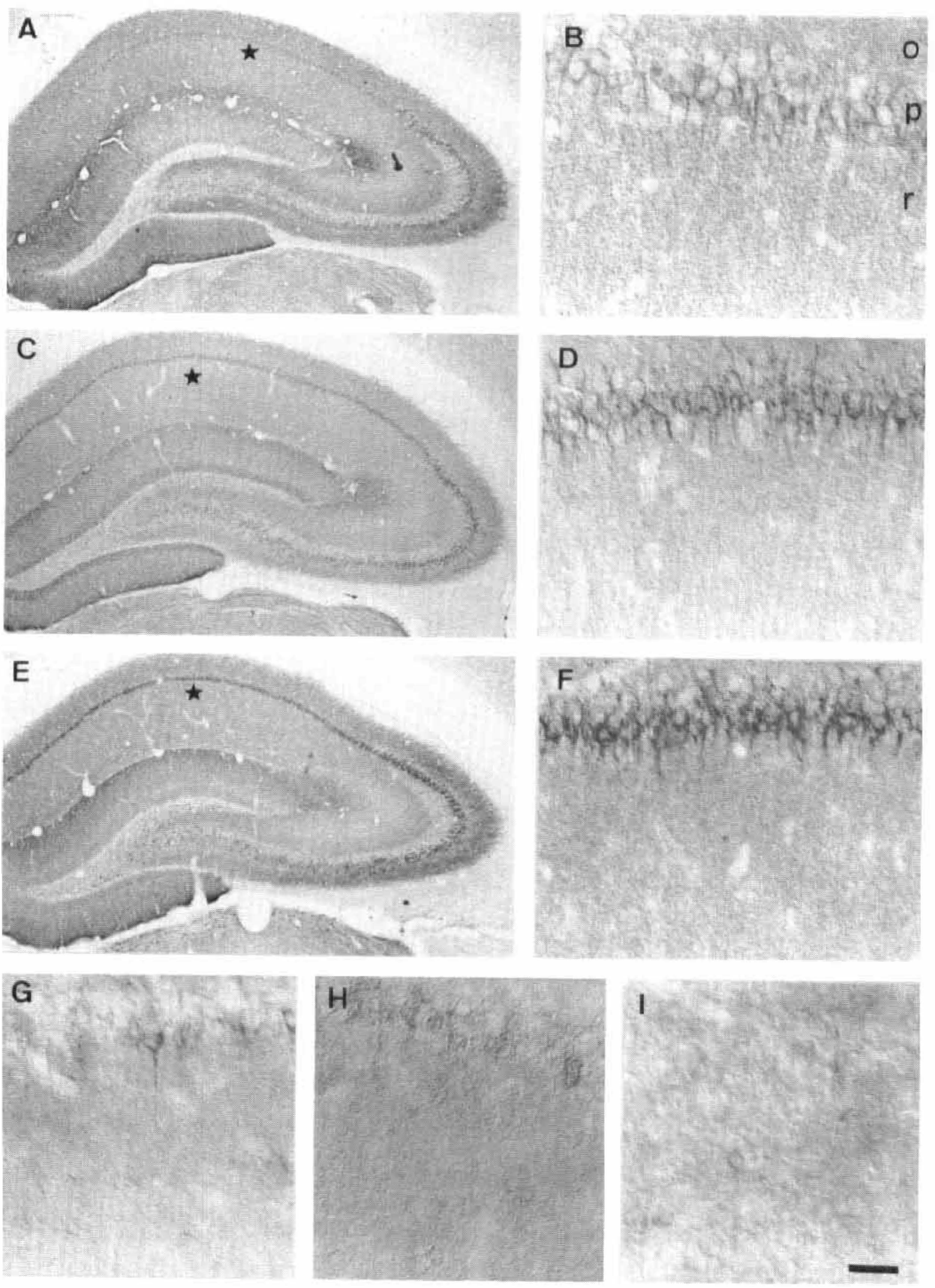
Immunolocalization of the glutamate transporter EAAC1 in the hippocampus of control animals
In addition to the changes of EAAC1 expression in the hippocampus, we also observed an increase of EAAC1 immunoreactivity with the same temporal profile in ischemic-sensitive regions such as layer V of the parietal cerebral cortex (Fig. 2) and the posterodorsal thalamus (Fig. 1E). In the parietal cerebral cortex from control animals, layer V pyramidal cell bodies and the surrounding neuropil were moderately stained with the EAAC1 antiserum, with weaker staining being observed throughout the remaining layers (Figs. 2A and 2B). These findings are in accordance with those reported previously (Rothstein et al., 1994). Ischemic insult caused a strong increase in EAAC1 immunoreactivity in layer V pyramidal neurons after 8 hours and 1 day of reperfusion (Fig. 2C to Fig. 2F). By 3 days after ischemia, EAAC1 levels in layer V were comparable to those in the parietal cerebral cortex of control animals (Figs. 2G and 2H) and remained like that at the longer postlesion periods examined (data not shown). In addition, a light increase in EAAC1 immunoreactivity was observed in cells, possibly pyramidal neurons, located in cortical cell layers II and VI (Fig. 2C to Fig. 2F).
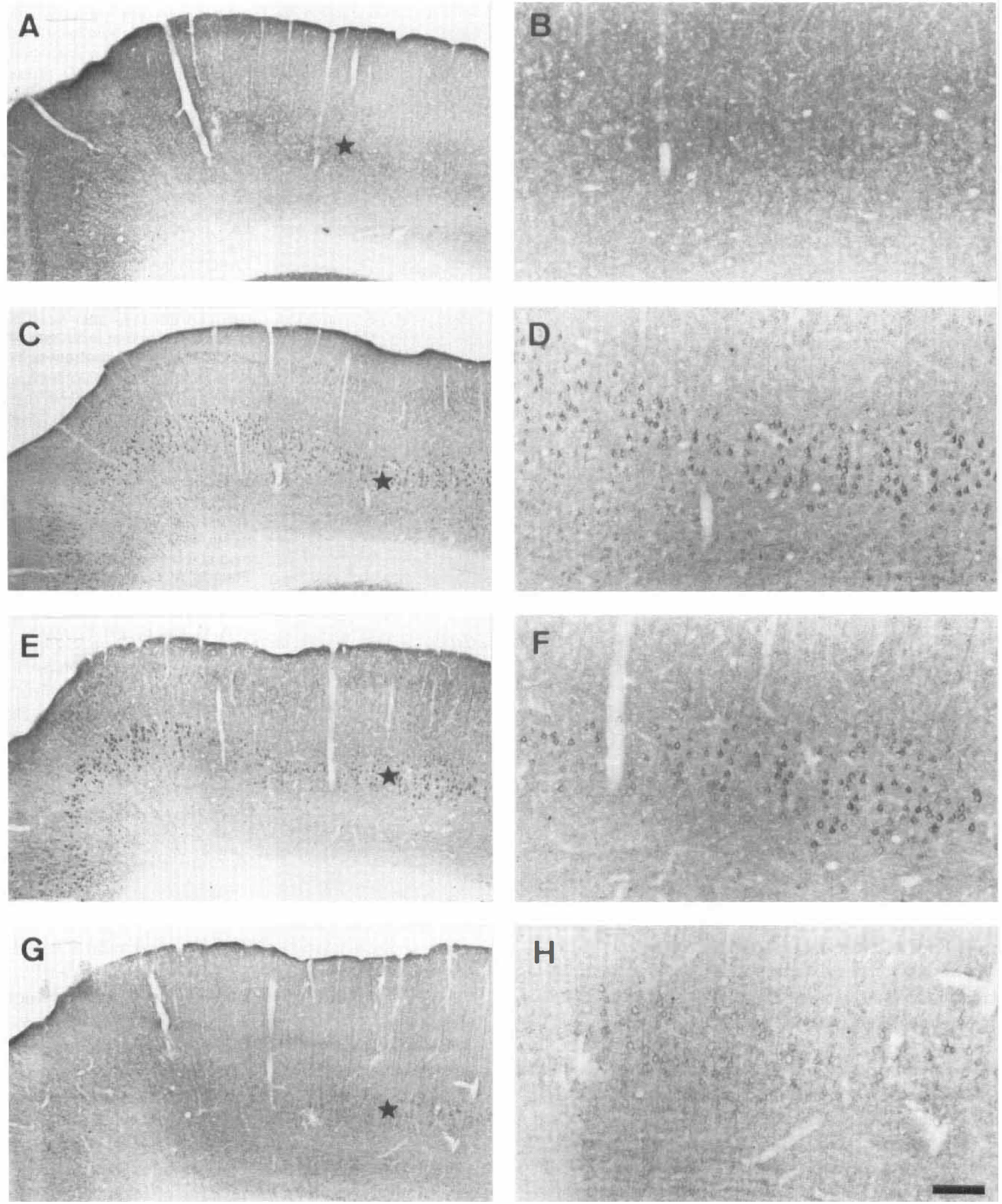
Immunolocalization of the glutamate transporter EAAC1 in the parietal cerebral cortex of control animals
Expression of the glutamate transporters GLAST and GLT-1 is largely unaltered after ischemia
We also investigated putative changes in the tissue distribution of the two major glial glutamate transporters GLAST and GLT-1 after ischemia. In control animals, GLAST and GLT-1 were located principally in the neuropil throughout the hippocampus (Figs. 3A and 3D, respectively), which is consistent with the idea of their presence in astroglial processes. Astroglial somata at light microscopic level were rarely stained, as previously described (Rothstein et al., 1994). In contrast to the neuronal glutamate transporter EAAC1, no major changes in the expression of GLAST and GLT-1, compared with controls, were observed in the hippocampus at the reperfusion stages examined (Figs. 3B, 3C, 3E, and 3F, respectively). Similarly, expression of these transporters in the cerebral cortex was unaltered after ischemia (data not shown). However, astrocytic cell bodies within the neuropil in CA1 became lightly immunostained after 3 days of recirculation (Figs. 3C and 3F).
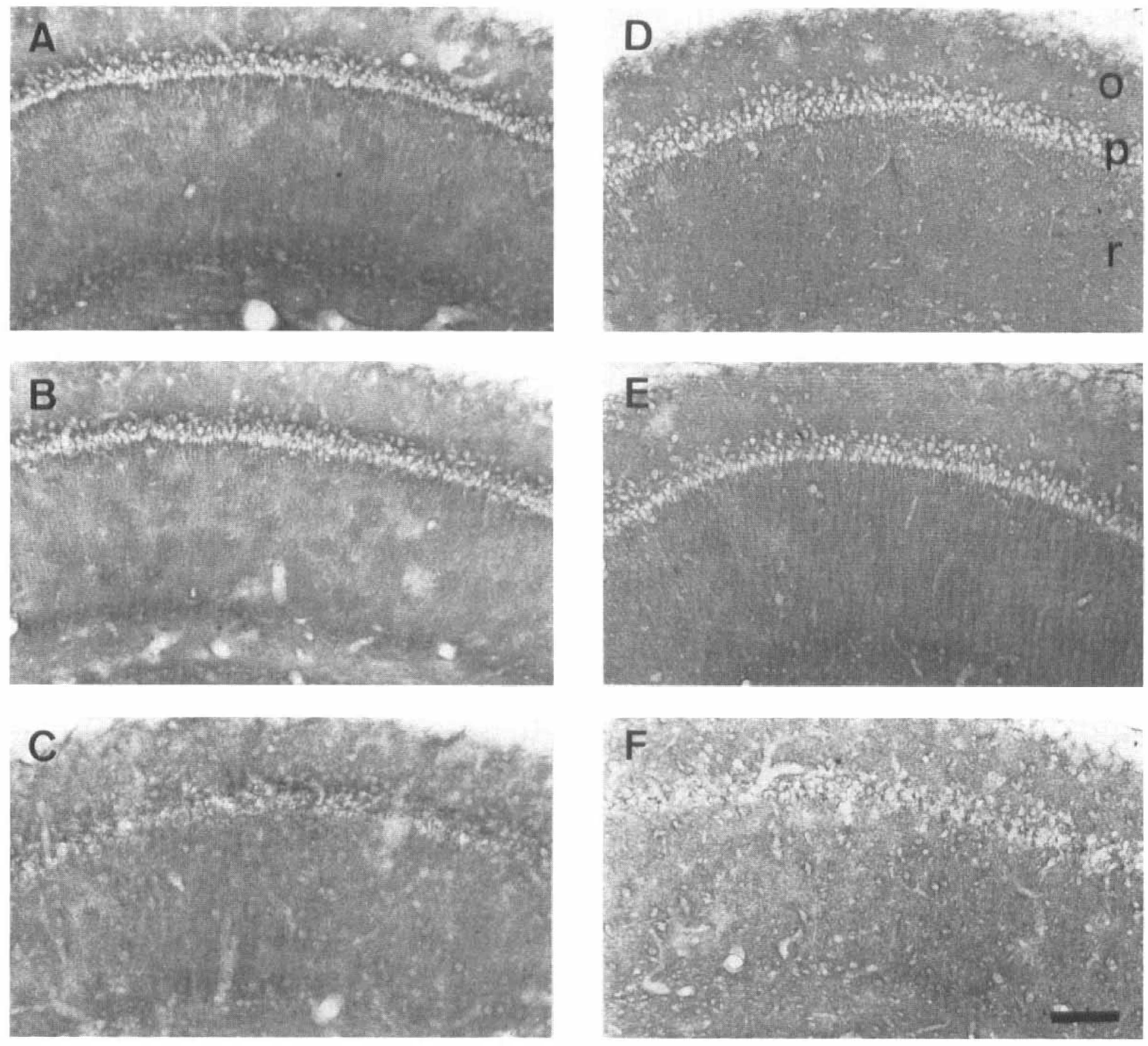
Immunolocalization of the glutamate transporters GLAST
Overall content of EAAC1, but not that of GLAST and GLT-1, in the CA1 is altered after ischemia
We next performed an immunoblotting analysis to estimate putative changes in the tissue content of EAAC1, GLAST, and GLT-1 in protein homogenates of the CA1 region after ischemia. Each antiserum labeled proteins of similar sizes to those described earlier (Rothstein et al., 1994; Domercq and Matute, 1999; Domercq et al., 1999). Thus, antisera to EAAC1, to GLAST, and to GLT-1 recognized a major peptide of about 69, 65, and 73 kDa, respectively, in all samples examined (not shown).
To quantify these findings, we first established the experimental parameters under which our immunoblot assays linearly detected increasing amounts of the corresponding transporter. It was thus determined that the amount of protein content in the homogenate that yielded a linear signal was up to around 15 μg per lane. Thus, we carried out a densitometric analysis of immunoblots (10 μg per lane of protein loaded). As expected from the localization studies, the levels of the GLAST and GLT-1 transporters did not show any significant change, compared with controls, at any of the postlesion stages examined (Fig. 4). In contrast, the levels of the EAAC1 transporter were augmented, compared with controls, after the initial postischemia stages and subsequently diminished (Fig. 4).
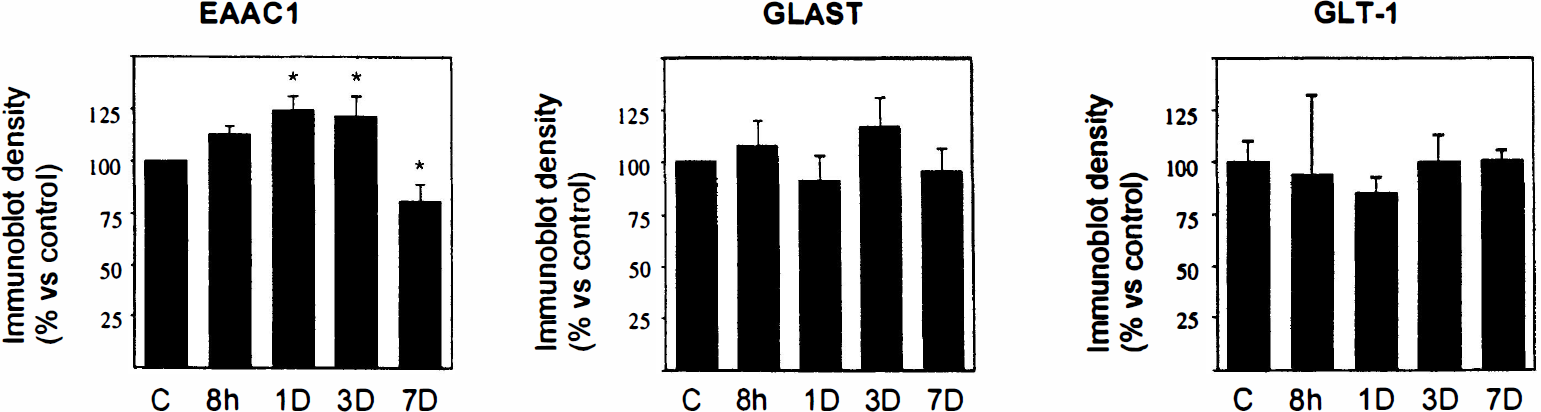
Histogram representing the densitometric analysis of immunoblots of glutamate transporters in CA1 control and postischemic protein homogenates (10 μg per lane). Notice that changes are statistically significant only for EAAC1. The ratio between transporter expression and total protein was calculated to correct differences in the amount of protein applied to each lane. Values represent the mean ± SD (n = 3), and statistics were carried out with analysis of variance and Fisher's post hoc analysis. *P < 0.05.
Number of EAAC1+ cells in subcortical white matter increases after ischemia
In addition to the presence of EAAC1 in synaptic regions, we previously observed that this transporter is expressed in cells of the oligodendroglial lineage, which are located in brain white matter areas (Domercq and Matute, 1999) and in the optic nerve (Domercq et al., 1999). Accordingly, antibodies to EAAC1 labeled immature-like cells of the subcortical white matter in control animals (Fig. 5A′ and Fig. 5B′) and after ischemia. All EAAC1+ cells were double-labeled with antibodies to GST-Yp+ (Fig. 5A), a marker of immature oligodendrocytes (Gensert and Goldman, 1996). Likewise, cells expressing EAAC1 were stained with antibodies to the NG-2 chondroitin sulfate proteoglycan, a marker of oligodendroglial progenitors (Nishiyama et al., 1996).
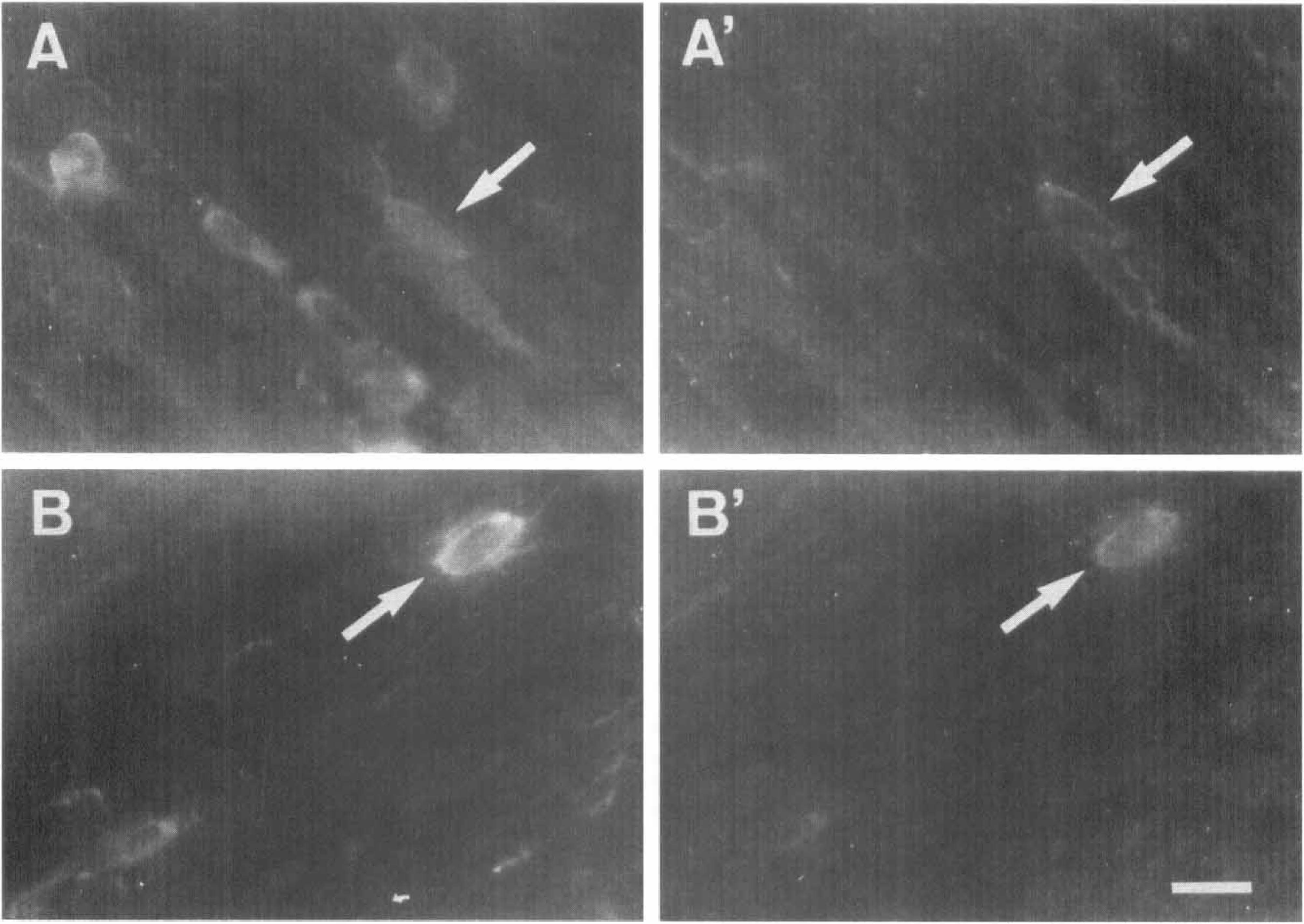
A subpopulation of immature oligodendrocytes express the glutamate transporter EAAC1. Morphologic appearance of cells belonging to the oligodendroglial lineage in control rat subcortical white matter, as revealed with antibodies to glutathione-S-transferase Yp subtype
Subsequently, we examined whether the number of EAAC1+ cells in subcortical white matter might be altered after ischemia by cell counting analysis of EAAC1+ oligodendroglial progenitor cells at 1 to 28 days after ischemia. The results show an increase in the density of EAAC1+ cells after reperfusion with a temporal profile, which was different from that described for EAAC1+ neurons in the hippocampus and parietal cortex (Table 1, Fig. 6). Thus, the density of EAAC1+ cells was higher after 3 postlesion days and reached a maximum (142% versus control) at 28 days after ischemia. No evidence of tissue shrinkage in the subcortical white matter was observed at the postischemic time periods examined, indicating that the higher density of EAAC1+ cells after ischemia corresponds to an increase in the total number of cells expressing this transporter. In contrast, no significant changes in the density of GLAST+ and GLT-1+ cells in the subcortical white matter were observed after lesion.
Quantitative analysis of EAAC1+ cells in periventricular subcortical white matter

Values represented mean ± SD for n = 3 independent experiments. Statistics were performed with analysis of variance and Fisher's post hoc analysis.
P < 0.05;
P < 0.01.
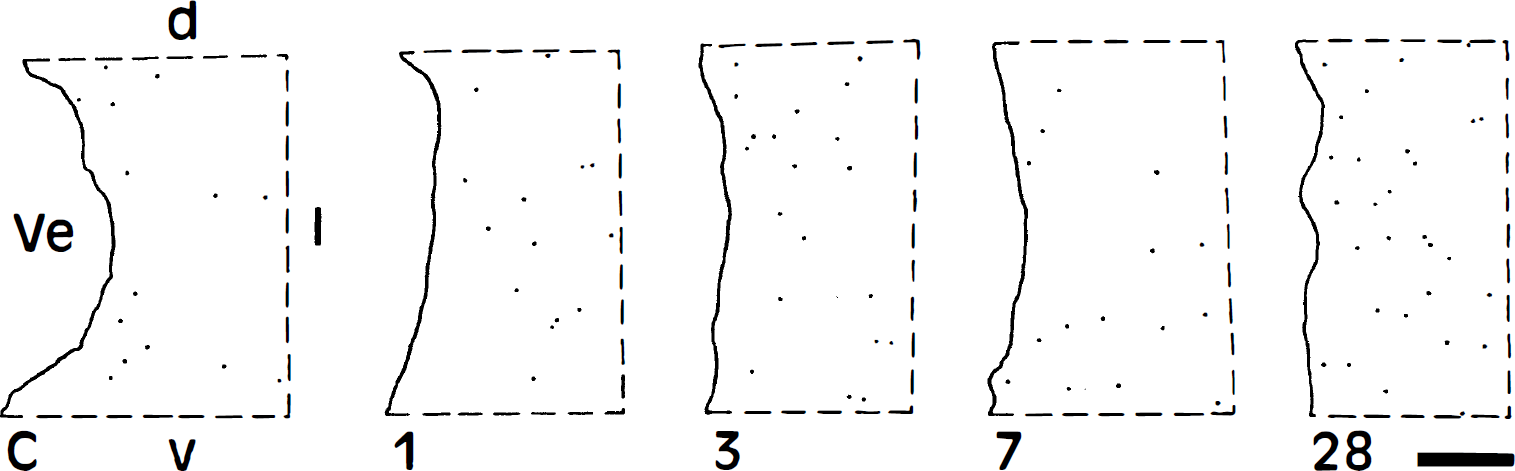
Drawings displaying the distribution of EAAC1+ cells in a portion of the periventricular subcortical white matter of representative immunostaining experiments using control brain (C) and at 1, 3, 7, and 28 days after ischemia. Ve, lateral ventricle; d, dorsal; l, lateral; v, ventral. Bar = 200 μm.
DISCUSSION
This study shows that after transient forebrain ischemia, there is a rapid increase in EAAC1 expression in pyramidal neurons of the hippocampal CA1 region and of the parietal cerebral cortex. In addition, we found that the ischemic insult causes a relatively slow increase in the number of EAAC1+ oligodendroglial progenitors cells in the subcortical white matter. These changes in glutamate transporter expression are specific for EAAC1, since the levels of GLAST and GLT-1 remained largely constant.
Increases in EAAC1 levels in CA1 pyramidal neurons occurred at early postischemic stages and abruptly declined as most cells in the corresponding cell layer died. Therefore, it was not possible to follow up the temporal profile and time course of these changes in EAAC1 expression in these cells beyond the third postlesion day. However, the fact that many pyramidal cells in the neocortex survive the ischemic insult (Pulsinelli et al., 1982) allowed us to observe that EAAC1 returned to basal levels at 3 days after ischemia and remained so thereafter. It is thus likely that the increase in the neuronal glutamate transporter EAAC1 at the early postischemic stages is triggered by the elevation of the extracellular glutamate concentration occurring during transient ischemia (Benveniste et al., 1984, 1989). Alternatively, reactive glial cells may initiate or contribute to the observed changes in EAAC1. Interestingly, microglial cells respond rapidly to ischemia, with a time course (Morioka et al., 1991) partially overlapping that of EAAC1 up-regulation. The molecular mechanisms by which the EAAC1 expression is upregulated may involve the activation of receptors coupled to tyrosine kinase signaling pathways, as demonstrated in vitro (Davis et al., 1998; Davis and Robinson, 1998).
A similarly rapid increase in EAAC1 mRNA expression has been described in hypoglossal motor neurons after nerve injury (Kiryu et al., 1995). On the other hand, it has been recently described that EAAC1 mRNA levels are virtually unaltered in the gerbil CA1 region 1 day after 3 minutes of global forebrain ischemia (Fujita et al., 1999). These findings, together with ours, suggest that the EAAC1 protein increase after ischemia may be post-transcriptionally regulated. However, notice that the experimental conditions used in both studies are not directly comparable and, therefore, other explanations cannot be excluded.
The results obtained by immunoblot indicate that after ischemia, the overall levels of EAAC1 in CA1 largely parallel the observed increase of transporter levels in the soma and proximal dendrites of pyramidal cells within this area. Thus, these findings reflect a significant increase of EAAC1 in a well-defined subset of cells that result in a global increase of this protein in the CA1 area. In addition, our observations in the parietal cerebral cortex suggest that a similar process may occur in other ischemia-sensitive regions.
Our results indicate that oligodendroglial progenitor cells (GST-Yp+ and NG-2+) in subcortical white matter expressed the EAAC1 glutamate transporter. Consistent with these results, previous evidence indicates that EAAC1 was located in glial cells of these areas (Kiryu et al., 1995; Conti et al., 1998) although the cell types involved were not completely elucidated. Moreover, recent findings in the corpus callosum indicate that immature oligodendroglia express EAAC1 (Domercq and Matute, 1999).
Intriguingly, the number of cells expressing EAAC1 in subcortical white matter increases at 3 days after ischemia and was maximal at 28 days of reperfusion. Several alternative explanations may account for this observation, such as an increase in the number of EAAC1+ oligodendrocyte progenitor cells from proliferation triggered by ischemia. This idea is supported by the fact that pairs of EAAC1+ cells were occasionally observed. Moreover, it is well established that resting adult oligodendrocyte progenitors can divide in the presence of various growth factors (Shi et al., 1998). The fact that both postischemic astrocytes and microglia can upregulate the synthesis of numerous neurotrophic factors (Lindvall et al., 1992; Takeda et al., 1993; Gottlieb and Matute, 1999) points to these cells as the putative source of trophic activity regulating the proliferation of adult oligodendroglial progenitor cells. Interestingly, after ischemia, the increases in the number of progenitors coincide with the periods of maximal microgliosis (Morioka et al., 1991) and astrogliosis (Petito et al., 1990). This suggests that microglia may control the proliferative activity of these cells at the early postischemia stages, whereas the later proliferative wave may be driven by astrocytes. Alternatively, it is possible that de novo expression of the transporter in the preexisting oligodendrocyte progenitor cell population occurs, although no evidence supports this hypothesis. Finally, it is unlikely that EAAC1+ cells in the postischemic subcortical white matter are not oligodendrocyte progenitors, since we did not observe expression of this transporter in other cell types after double-labeling experiments (data not shown).
The increased size of the immature oligodendroglial population may subserve a repair process to compensate for the loss of mature oligodendrocytes occurring on ischemia. Indeed, white matter areas are vulnerable to ischemic brain injury (Follis et al., 1993; Pantoni et al., 1996), a feature which may be partially related to oligodendroglial excitotoxic cell death mediated by glutamate receptors (Yoshioka et al., 1996; Matute et al., 1997; McDonald et al., 1998). Thus, glutamate release by reversed operation of glutamate transporters at Ranvier nodes in response to the ionic alterations described in white matter regions after ischemic and anoxic insults could induce oligodendroglial death (reviewed in Stys, 1998). Under this condition, oligodendroglial progenitor cell proliferation could replenish damaged oligodendrocytes.
Our results show that alterations in glutamate transporter expression after ischemia involve mainly EAAC1, since we did not observe significant changes in the levels of GLAST and GLT-1 transporters using immunohistochemical and immunoblot analyses. Consistently, it has been observed in the gerbil that GLAST mRNA levels are largely unchanged during the first week postischemia (Fujita et al., 1999). In contrast, GLT-1 mRNA levels were lowered in this ischemia model (Fujita et al., 1999). Several features, including differences in the ischemia model used, posttranscriptional regulation of the transporter, and compensatory expression of noncharacterized GLT-1 splice variants recognized by the antibody used, may account for the apparent discrepancy between the two studies. Indeed, the cRNA probe used for hybridization by Fujita et al. (1999) does not recognize the region encoding the C-terminus of the GLT-1 protein, the domain to which the antibody used in the current study was raised (Rothstein et al., 1994, 1996).
Thus, although the expression of GLT-1 in vitro is regulated by neuron-secreted factors (Swanson et al., 1997) or that of GLAST by ionotropic glutamate receptor activation (Gegelashvili et al., 1996), the molecular alterations after ischemia are not sufficient to induce these changes. For instance, the upregulation of GLAST in vitro requires prolonged treatment (7 days) with glutamate or other agonists (Gegelashvili et al., 1996). Since the increase in extracellular glutamate from reversion of transporters is transitory (Szatkowsky and Attwell, 1994), it is possible that the ischemia-associated stimulus is not sufficiently prolonged to induce changes in glial glutamate transporter expression. Accordingly, a slight reduction in hippocampal GLT-1 expression by 3 hours after transient forebrain ischemia, but not at 24 hours after ischemia, has been described (Torp et al., 1995).
In summary, we show evidence for an upregulation of EAAC1 in a defined population of cells within ischemia-sensitive areas, together with an increase in the number of EAAC1+ oligodendroglial progenitor cells in subcortical white matter. Thus, increased expression of EAAC1 may subserve neuroprotective mechanisms occurring after ischemia.
Footnotes
Acknowledgements
The authors thank Dr. J. D. Rothstein for the gift of polyclonal antibodies to the glutamate transporters examined, Dr. Levine for providing antibodies to the NG-2 chondroitin sulphate, and D. J. Fogarty for reviewing the manuscript.