
Abstract
We previously reported that during pro-estrus (high endogenous estrogen levels), brain damage after middle cerebral artery occlusion (MCAO) was reduced in stroke-prone spontaneously hypertensive rats (SHRSP) but not in normotensive Wistar Kyoto rat (WKY). In the present study, we examined the effect of exogenous estrogen on brain damage after MCAO in SHRSP and WKY. A 17β-estradiol (0.025mg or 0.25mg, 21 day release) or matching placebo pellet was implanted into ovariectomized WKY and SHRSP (3 to 4 months old) who then underwent distal diathermy-induced MCAO 2 weeks later. Plasma 17β-estradiol levels for placebo and 17β-estradiol groups were as follows: WKY 0.025 mg 16.4 ± 8.5 (pg/mL, mean ± SD) and 25.85 ± 12.6; WKY 0.25 mg 18.2 ± 9.0 and 69.8 ± 27.4; SHRSP 0.25 mg 20.7 ± 8.8 and 81.0 ± 16.9. In SHRSP, infarct volumes at 24 hours after MCAO were similar in placebo and 17β-estradiol groups: SHRSP 0.025 mg 126.7 ± 15.3 mm3 (n = 6) and 114.0 ± 14.1 mm3 (n = 8) (not significant); SHRSP 0.25 mg 113.5 ± 22.3 mm3 (n = 8) and 129.7 ± 26.2 mm3 (n = 7) (not significant), respectively. In WKY, 17β-estradiol significantly increased infarct volume by 65% with 0.025mg dose [36.1 ± 20.7 mm3 (n = 8) and 59.7 ± 19.3 mm3 (n = 8) (P = 0.033, unpaired t-test)] and by 96% with 0.25 mg dose [55.9 ± 36.4 mm3 (n = 8) and 109.7 ± 6.7 mm3 (n = 4) (P = 0.017)]. Thus, 17β-estradiol increased stroke damage in normotensive rats with no significant effect in stroke-prone rats. Despite being contrary to our hypothesis, our findings add substance to the recently reported negative effects of 17β-estradiol in clinical studies.
Animal stroke model studies have reported 17β-estradiol to be neuroprotective (Hurn and Macrae, 2000). 17β-estradiol administration to ovariectomized rats before transient middle cerebral artery occlusion (MCAO) (Rusa et al., 1999; Simpkins et al., 1997) and permanent MCAO (Dubal et al., 1998) significantly reduces the amount of brain damage. Post-MCAO treatment of 17β-estradiol has also been shown to be neuroprotective (Yang et al., 2000). Such neuroprotective effects of 17β-estradiol in experimental stroke do not appear to be gender-specific, given that Toung et al. (1998) reported 17β-estradiol neuroprotection in male rats. In addition, these effects do not appear to be species specific, given that Culmsee et al. (1999) has shown estrogen-induced neuroprotection in mice. We have shown that cycling rats in pro-estrus, when 17β-estradiol levels are high, exhibit less brain damage after distal permanent MCAO than in met-estrus, when 17β-estradiol levels are low (Carswell et al., 2000). This was found in a stroke sensitive strain, the stroke-prone spontaneously hypertensive rat (SHRSP), but not in their normotensive counterpart, the Wistar Kyoto rat (WKY). In addition, the neuroprotective effects of 17β-estradiol extend in other models of brain damage such as global ischemia in mice (Horsburgh et al., 2002) and rats (Wang et al., 1999) and neurotoxic lesions to the hippocampus in rats (Azcoitia et al., 1998).
The clinical evidence for beneficial effects of estrogen in human stroke is less convincing. Whereas some observational studies have shown hormone replacement therapies (HRT) to decrease stroke risk (Finucane et al., 1993), other observational studies as well as randomized placebo-controlled clinical trials have shown estrogen replacement to increase stroke risk and mortality (Viscoli et al., 2001; Wilson et al., 1985).
The present study uses stroke sensitive (SHRSP) and normotensive (WKY) rats to examine whether 17β-estradiol replacement at low and high physiologic doses reduces brain damage in the diathermy model of distal MCAO and to examine if SHRSP would derive more benefits from 17β-estradiol than WKY rats. SHRSP have genetically determined hypertension and increased sensitivity to experimental stroke compared with WKY rats (Carswell et al., 1999; Coyle and Jokelainen, 1983; Jeffs et al., 1997) and an increased frequency of spontaneous stroke (Okamoto et al., 1974), the pathology of which is similar to strokes in humans (Yamori et al., 1976).
METHODS
All experiments were performed according to the UK Home Office Guidelines, with the approval of the University of Glasgow Ethical Review Panel, and subject to the Animals (Scientific Procedures) Act, 1986. Breeding and housing of SHRSP and WKY rats has previously been described (Clark et al., 1996). Female SHRSP and WKY (3 to 4 months of age; WKY 205 ± 10 g; SHRSP 179 ± 11 g) were anesthetized with halothane (induction 5%; maintenance 1% to 2% in 100% oxygen using a face mask) and injected with 0.02 mL carprofen (Rimadyl, 50 mg/mL; Pfizer Animal Health, Kent, UK) for postoperative analgesia before bilateral ovariectomy. A 21-day release 17β-estradiol or placebo pellet (Innovative Research of America) was then implanted subcutaneously at the nape of the neck. Four groups of females were used in each strain, receiving either 0.025 mg 17β-estradiol (SHRSP n = 8; WKY n = 8) with matching placebo (SHRSP n = 6; WKY n = 8) or 0.25 mg 17β-estradiol (SHRSP n = 7; WKY n = 4) with matching placebo (SHRSP n = 8; WKY n = 8).
Two weeks later, the animals underwent MCAO. Anesthesia was induced by 5% halothane in oxygen-nitrous oxide (30:70) and maintained by intubation and ventilation with 1% to 2% halothane. A 2-mm distal segment of the left MCA was occluded by electrocoagulation using a modified version of the original Tamura model (Tamura et al., 1981) to produce a predominantly cortical lesion (Carswell et al., 2000). Physiologic variables were monitored throughout the period of anesthesia and at 24 hours after MCAO as previously described (Carswell et al., 1999). 24 hours after MCAO, blood was collected for measurement of plasma 17β-estradiol levels, and animals were perfusion fixed with 4% paraformaldehyde in 50 mM phosphate buffer. Brains were dehydrated and processed for paraffin embedding, and 6μm coronal sections were cut and stained with hematoxylin-eosin. The infarct was delineated onto line diagrams at eight coronal planes throughout the MCA territory to correct for brain swelling, and the infarct areas were measured by image analysis (MCID, Imaging Research, St. Catherines, Ontario, Canada). The volume of infarction for each brain was derived by integration of areas of infarction over the eight coronal levels measured with end-points of 12.5 mm anterior and 0.05 mm posterior to the interaural line (Osborne et al., 1987). Student's unpaired two-tailed t-test was used for statistical analysis of infarct volumes between estrogen and matching placebo groups. Plasma 17β-estradiol levels were measured either by a radioimmunoassay (Coat-a-Count, Diagnostics Products Corporation) or an Enzyme Linked Immunoassay (ELISA, DRG Instruments, Germany) after extraction of 17β-estradiol from the rat plasma and reconstitution into human serum. Plasma levels from the different treatment groups were compared using Student's unpaired two-tailed t-tests. Low dose samples were analyzed first, and because of technical difficulties with the radioimmunoassay analysis of SHRSP samples, the ELISA (which had a lower detection limit than the radioimmunoassay) was used for analysis of all plasma levels thereafter (comprised of the higher dose samples). Body weights, measured during the 2-week period between ovariectomy and MCAO, in 17β-estradiol and placebo groups were compared by Student's unpaired two-tailed t-test. Data are presented as mean ± SD.
RESULTS
Physiologic parameters and plasma 17β-estradiol levels
Physiologic parameters for the experiments in the present study are illustrated in Table 1. All physiologic variables were maintained within normal limits under anesthesia. Both anesthetized and conscious (24 hours after MCAO) mean arterial blood pressures (MAP) were higher in SHRSP than in WKY, as expected. Plasma 17β-estradiol levels were higher in estrogen-treated groups than in placebo groups.
Plasma 17 β-estradiol levels and physiological parameters
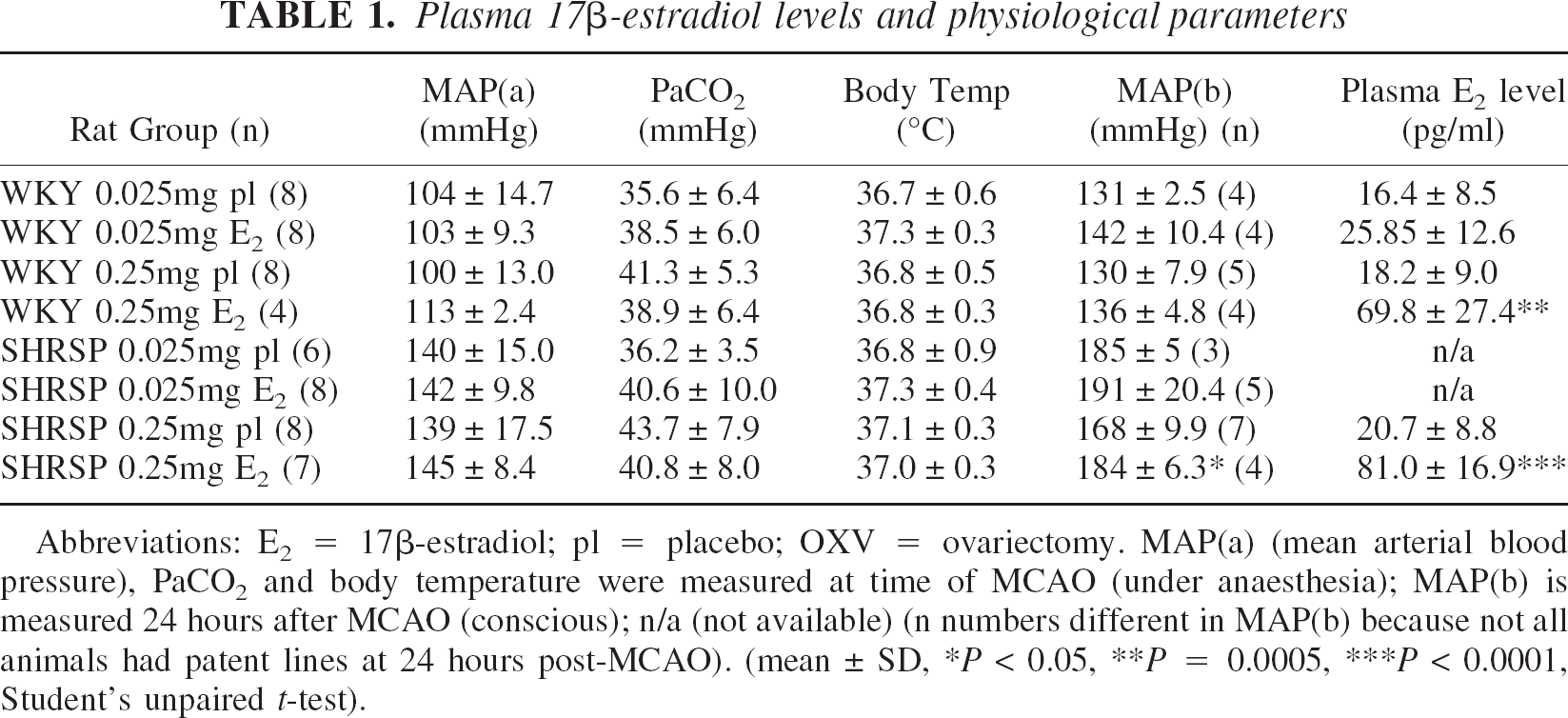
Abbreviations: E2 = 17β-estradiol; pl = placebo; OXV = ovariectomy. MAP(a) (mean arterial blood pressure), PaCO2 and body temperature were measured at time of MCAO (under anaesthesia); MAP(b) is measured 24 hours after MCAO (conscious); n/a (not available) (n numbers different in MAP(b) because not all animals had patent lines at 24 hours post-MCAO). (mean ± SD, ∗P < 0.05, ∗∗P = 0.0005, ∗∗∗P < 0.0001, Student's unpaired t-test).
Ischemic damage
WKY rats treated with 0.025 mg 17β-estradiol had significantly larger infarct volumes (increased by 65%) compared with WKY rats treated with matching placebo (59.7 ± 19.3 mm3 and 36.1 ± 20.7 mm3, respectively, P = 0.033) (Fig. 1A). The same dose of 17β-estradiol in SHRSP rats did not significantly affect the volume of infarction (114 ± 14.1 mm3 for 17β-estradiol and 126.7 ± 15.3 mm3 for placebo, not significant) (Fig. 1A). A 10-fold higher dose of 17β-estradiol significantly increased infarct volume in WKY rats by 96% compared with the matching placebo group (109.7 ± 6.7 mm3 and 55.8 ± 36.4 mm3 respectively, P = 0.0168) (Fig. 1B) but not in SHRSP rats (129.7 ± 26.2 mm3 and 113.5 ± 22.3 mm3 respectively, not significant) (Fig. 1B). Fig. 2A and Fig. 2C illustrate a clear increase in infarction induced by 17β-estradiol in WKY rats throughout the MCA territory, with the least difference at the rostral and caudal extents of the MCA territory. However, in the SHRSP, there is no clear difference in the infarct areas throughout the MCA territory between 17β-estradiol and placebo groups at either dose (Fig. 2B and Fig. 2D).
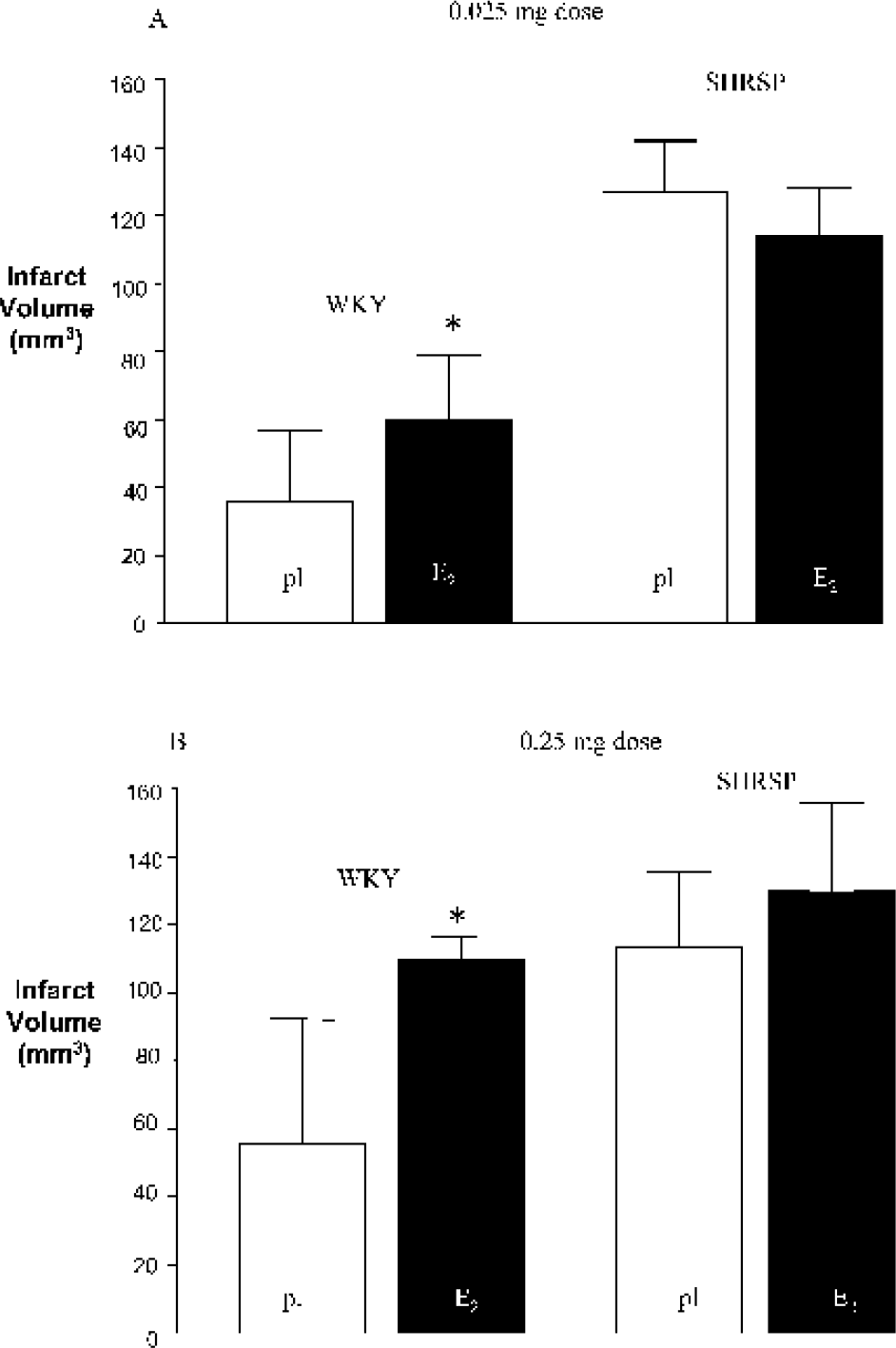
Volumes of infarct (mm3) for (A) 0.025 mg dose and (B) 0.25 mg dose of E2 and matching pl-treated WKY and SHRSP. ∗ P < 0.05 versus placebo. E2, 17β-estradiol; pl, placebo; WKY, Wistar Kyoto rats; SHRSP, stroke-prone spontaneously hypertensive rats.
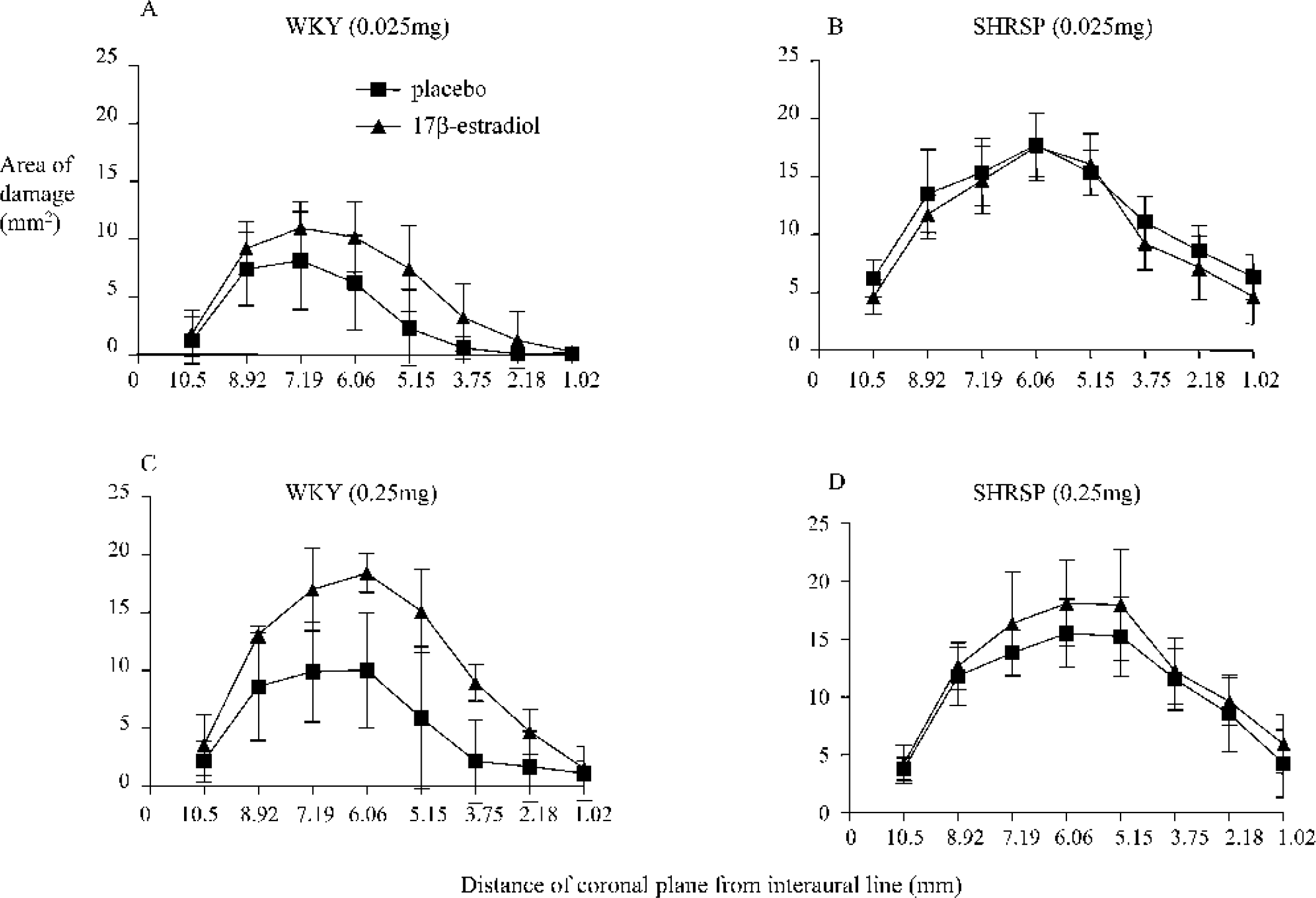
Areas of infarct (mm2) over eight coronal planes for 0.025 mg dose in (A) WKY and (B) SHRSP and for 0.25 mg dose of E2 and matching pl-treated in (C) WKY and (D) SHRSP. E2, 17β-estradiol; pl, placebo; WKY, Wistar Kyoto rats; SHRSP, stroke-prone spontaneously hypertensive rats.
Body weights
Fig. 3 illustrates change in body weight over the 2-week treatment period between ovariectomy and MCAO. The increase in body weight over the 2 weeks was significantly less in animals treated with 17β-estradiol than in the placebo-treated animals in both strains at the 0.025 mg dose. At the 0.25 mg dose, 17β-estradiol induced a weight loss over the 2 weeks after ovariectomy, whereas the matching placebo animals gained weight. The change in body weight after ovariectomy did not correlate significantly with infarct volume in any of the groups [WKY 0.025 mg 17β-estradiol: r2 = 0.02 (P =0.7) and placebo: r2 = 0.0006 (P = 1.0); SHRSP 0.025 mg 17β-estradiol: r2 = 0.05 (P = 0.6) and placebo: r2 = 0.08 (P = 0.6); WKY 0.25 mg 17β-estradiol: r2 = 0.4 (P = 0.4) and placebo: r2 = 0.04 (P = 0.6); SHRSP 0.25 mg 17β-estradiol: r2 = 0.4 (P = 0.1) and placebo: r2 =0.04 (P = 0.6)].
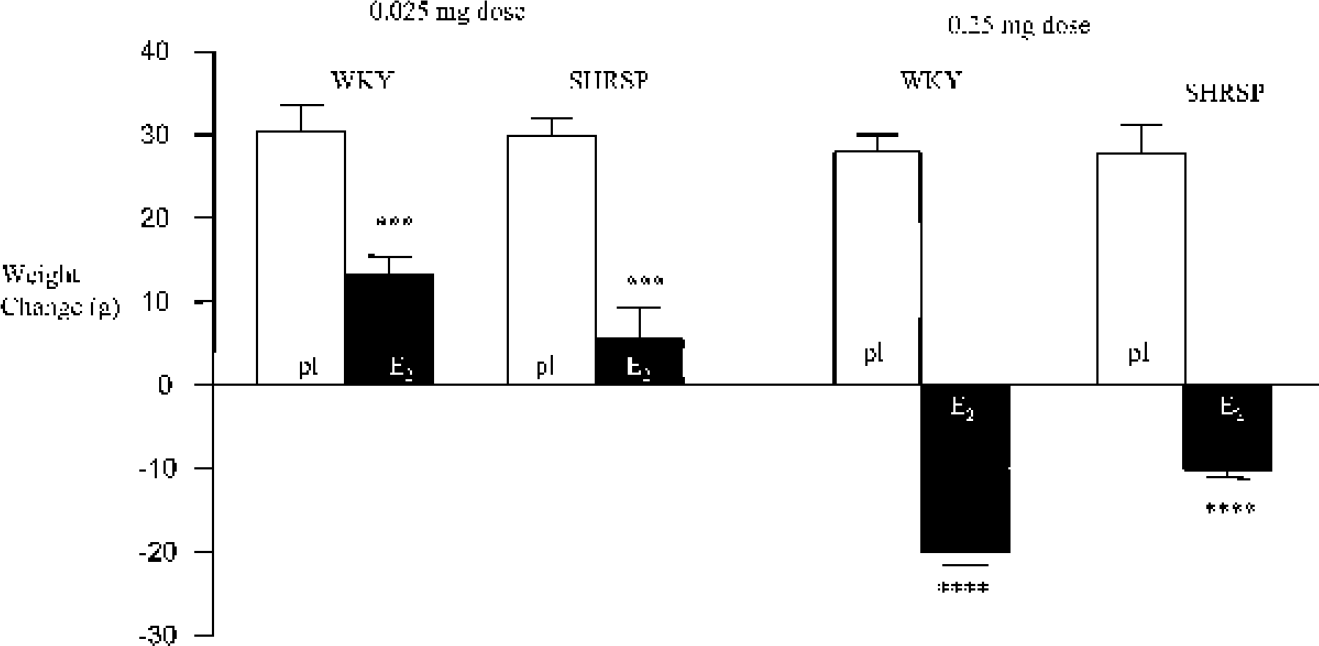
Change in body weight during 2-week treatment period between ovariectomy and MCAO in E2 and matching pl-treated WKY and SHRSP. ∗∗∗ P < 0.001, ∗∗∗∗ P < 0.0001 versus matching pl treatment. MCAO, middle cerebral artery occlusion; E2, 17β-estradiol; pl, placebo; WKY, Wistar Kyoto rats; SHRSP, stroke-prone spontaneously hypertensive rats.
DISCUSSION
The key findings of the present study are that estrogen causes an increase in infarct volume after distal MCAO in normotensive rats and, at the doses used, has no significant effect on infarct volume in the SHRSP. Parameters such as age, body temperature, blood pressures, and arterial blood gases were similar in estrogen- and placebo-treated WKY. Body weight was reduced by estrogen, possibly mediated by deceased hypothalamic neuropeptide Y release (Bonavera et al., 1994), but this did not correlate with infarct size. These parameters therefore do not account for the increase in infarct size in WKY. In addition, brain swelling was corrected for by transcribing the distribution of infarcts onto line diagrams to measure infarct size. Thus the increase in infarct size in WKY is caused by a true exacerbation of the infarct volume and not simply to an increase in brain edema.
Despite being contrary to our hypothesis, these findings clearly demonstrate detrimental effects of estrogen after MCAO and substantiate earlier reports of deleterious effects of estrogen in global ischemia (Harukuni et al., 2001) and in human stroke (Rossouw et al., 2002; Viscoli et al., 2001). Additionally from our laboratory, using the same treatment regimen, Bingham et al. (unpublished observations, 2003) have demonstrated an increase in infarct volume after proximal diathermy MCAO in Lister Hooded rats, and Wallace et al. (unpublished observations, 2003) have shown an increase in infarct volume induced by the 0.25 mg dose of estrogen in the intraluminal thread (ILT) model of permanent focal ischemia in Sprague Dawley rats. Therefore, although the earlier literature reported beneficial influences of 17β-estradiol on ischemic damage, there is now a growing body of evidence supporting detrimental influences of the hormone in cerebral ischemia.
Differences in the study design between this study and others that report neuroprotection are likely to indicate reasons for the different influence of estrogen on brain injury after MCAO. First, the present study uses electrocoagulation of the distal MCAO followed by section of the artery (based upon Tamura et al., 1981), the most reliable of the current models to induce a permanent ischemic insult (Macrae, 1992). This model has not been used in any of the previous estrogen stroke studies. Most of the studies that report estrogen-mediated neuroprotection use the ILT model (Hurn and Macrae, 2000), which involves insertion of a thread to block blood flow at the origin of the MCA. One possibility is that the model of stroke used influences the balance between the beneficial and detrimental effects of estrogen. For example, regional and severity differences in ischemia and pathology exist after blocking the MCA by thread compared with electrocoagulation. Even if the thread is left permanently in place across the origin of the MCA, there is still a possibility of a small residual blood flow through the MCA, which could be improved by estrogen, unlike the electrocoagulation model. Furthermore, in the WKY, the distal diathermy model causes a small infarct relative to the ILT model, thus providing more opportunity for 17β-estradiol to increase the extent of ischemic damage. However, recent evidence from our laboratory of an increase in brain damage induced by the higher (0.25 mg) dose of 17β-estradiol in the permanent ILT model (Wallace et al., unpublished observations, 2003) does not support these speculations. Alternatively, the different mechanisms of injury in permanent versus transient ischemia may explain differential effects of estrogen. For example, transient ischemia models that incorporate reperfusion of the ischemic tissue may derive more benefit from vasodilatory and antioxidant effects of estrogen than permanent ischemia.
Clearly the timing, duration, dose, and formulation of estrogen administered and the plasma estrogen levels achieved are potential determinants of estrogen's influence upon stroke. Using normotensive animals, experimental studies in stroke have shown estrogen neuroprotection using pretreatment regimens similar in timing and duration to the present study (Dubal et al., 1998; Rusa et al., 1999). For purposes of comparison, physiologic estradiol levels during the rat estrous cycle are approximately 7 to 20 pg/mL in met-estrus, peaking to 50 to 88 pg/mL in pro-estrus (Nequin et al., 1979; Smith et al., 1975). In the present study, the 0.025 mg dose replaced 17β-estradiol within the met-estrus physiologic range, and the 0.25 mg dose replaced 17β-estradiol to values in the pro-estrus range. Rusa and colleagues (1999) induced transient ischemia using the ILT model and reported neuroprotection with a similar physiologic dose (0.025 mg 21-day release, plasma levels of 20 pg/mL), duration (7 to 16 days pretreatment) and formulation of 17β-estradiol as in the present study. However, neuroprotection was lost with a higher dose (0.1 mg dose, plasma levels of 46 pg/mL). In another neuroprotection study using the ILT model to induce permanent ischemia, Dubal et al. (1998) have reported effective neuroprotection at two doses (plasma levels of 10 pg/mL and 60 pg/mL) (7-day pretreatment). It is clear from these studies that the low dose used would be expected to exert neuroprotection, albeit the therapeutic range does appear to vary from laboratory to laboratory.
Concerning the SHRSP, this is the first study to investigate the effects of estrogen pretreatment on MCAO in SHRSP. Previously, Alkayed et al. (1998) showed that nonovariectomized SHRSP had smaller infarcts after MCAO than ovariectomized SHRSP, and our own studies revealed neuroprotection during the pro-estrus phase of the estrous cycle in the SHRSP (Carswell et al., 2000). Given that 17β-estradiol is the main ovarian and most biologically active estrogen, we expected 17β-estradiol to induce neuroprotection in SHRSP in the present study. The lack of benefit is unlikely to be explained by a type II error because the same model and number of animals were used as in Carswell et al. (2000). The difference in outcome may be because of the use of exogenous (synthetic) versus endogenous estrogen, the maintained, stable (14 days) 17β-estradiol levels versus fluxes (<24 hours) of cycling estrogens, or the absence of fluctuating progesterone levels.
Another possible factor for determining estrogen's influence upon stroke is the strain of rats used. Estrogen sensitivity could vary among strains because of different levels of estrogen receptors or estrogen binding proteins or because of different rates of estrogen breakdown. Wistar and Sprague Dawley rats have most commonly been used to investigate estrogen-induced neuroprotection (Dubal et al., 1998; Rusa et al., 1999; Simpkins et al., 1997), whereas WKY and SHRSP were used in the present study. However, estrogen-induced exacerbation of damage is not specific to WKY; we have recently confirmed exacerbation of stroke damage in Lister Hooded and Sprague Dawleys rats in our laboratory (unpublished observations, 2003). We have, however, shown a strain difference in estrogen's influence upon brain damage after MCAO in the present study in that 17β-estradiol causes detrimental effects in WKY but no significant effects in SHRSP. This could be indicative of an interaction between genetic background (strain) and estrogen, which points towards possible gene-hormone interactions. The lack of a detrimental effect of 17β-estradiol in the SHRSP may be associated with the preexisting genetically determined sensitivity to ischemic damage in the SHRSP, possibly giving us mechanistic insight into estrogen-induced exacerbation of damage.
With regard to potential mechanisms responsible for exacerbation of damage, estrogen has been shown to potentiate the NMDA receptor response. Estrogen increases hippocampal NMDA receptor agonist binding sites (Weiland et al., 1992), NMDA receptor-mediated EPSPs, and LTP (Foy et al., 1999). Such properties of estrogen may contribute to a detrimental influence in stroke. In line with increasing synaptic input, estrogen has been shown to transiently reduce hippocampal glutamic acid decarboxylase (GAD) and suppress GABAA-mediated inhibition of CA1 pyramidal cells (Rudick et al., 2001) 24 hours after acute treatment. However, to have any functional relevance to the present study, we need to first establish whether these effects of estrogen are evident in the cortex after experimental stroke after 14 days of continuous treatment.
Despite being contrary to our hypothesis, our findings add substance to the recently reported negative effects of estrogen in clinical studies. Whereas some observational studies have shown HRT to decrease the risk of human stroke (Finucane et al., 1993), others have shown HRT to increase (Wilson et al., 1985) or have no effect (Pedersen et al., 1997) upon human stroke risk, albeit patient selection bias in such studies may affect the results. A randomized, double-blind, placebo-controlled clinical trial of 17β-estradiol therapy in postmenopausal women who had recently had an ischemic stroke or transient ischemic attack has shown that estrogen increased risk of fatal stroke and worsened neurologic outcome after non-fatal stroke (Viscoli et al., 2001). A randomized trial of conjugated equine estrogen plus medroxyprogesterone in healthy postmenopausal women reported that overall health risks including an increase in stroke rate exceeded benefits from the combined treatment (Rossouw et al., 2002). Such effects of estrogen are in agreement with the recent study by Harukuni et al. (2001) showing that there are indeed deleterious effects of 17β-estradiol in a model of four-vessel global ischemia. Taken together with the present findings, it cannot always be assumed that estrogen will exert benefit.
CONCLUSION
Estrogen's overall effect on stroke is likely to be a combination of many complementary and opposing mechanisms exerted during the evolution of the infarct, thus making the role of estrogen in stroke very complex. It seems clear that, depending upon the local environment in the region of brain ischemia, estrogen may exhibit either beneficial or detrimental effects. There is a pressing need to determine the precise mechanisms influenced by estrogen in stroke before any more clinical trials are performed and because millions of women world-wide currently take estrogen in the form of replacement therapy or the contraceptive pill. We have provided evidence that estrogen can worsen stroke in an animal stroke model. These findings relate to the findings in human stroke and provide a valuable means for elucidating the detrimental mechanisms involved.
Footnotes
Acknowledgments
The authors thank Dr. Delyth Graham, Mrs. Lindsay Gallagher, Mrs. Joan Stewart, and Mrs. Margaret Stewart for assistance and technical expertise.