
Abstract
Advanced glycation endproducts (AGEs) accumulate on long-lived proteins, including β-amyloid plaques in Alzheimer's disease, and are suggested to contribute to neuronal dysfunction and cell death. We have investigated the effects of a model AGE upon glucose metabolism and energy production in a neuroblastoma cell line. AGEs decrease cellular ATP levels and increase glucose consumption and lactate production. All of the AGE-induced metabolic changes can be attenuated by antioxidants such as (R+)-α-lipoic acid and 17β-estradiol. These antioxidants may become useful drugs against (AGE-mediated) effects in neurodegeneration through their positive effects on cellular energy metabolism.
The amino groups of proteins, and particularly those on lysine residues, react nonenzymatically with reducing sugars. This posttranslational modification is termed nonenzymatic glycosylation, glycation, or Maillard reaction and leads, via reversible Schiff-base adducts, to the formation of Amadori products. Through subsequent oxidation and dehydration steps, a broad range of heterogeneous fluorescent and brown products is formed, called the advanced glycation endproducts (AGEs). These reactions cause protease-resistant cross-linking of peptides and proteins, leading to protein deposition and amyloidosis (Brownlee, 1995). Deposition of AGE-crosslinked insoluble protein aggregates is characteristic of beta-2-microglobulin (in dialysis-related amyloidosis) or islet amyloid polypeptide (in Type II diabetes mellitus) (Miyata et al., 1994b). In the brains of patients with Alzheimer's disease (AD), AGEs accumulate on β-amyloid plaques and in microglia and astrocytes in their vicinity (Sasaki et al., 1998). Because the average age of isolated plaques was determined to be around 14 years (Shapira et al., 1988), it is likely that AGEs progressively accumulate on these deposits over time to an extent comparable with synthetically prepared AGEs (Vitek et al., 1994). In summary, plaque proteins are likely to be as extensively modified by AGE as other long-lived proteins, including eye lens crystallin and β2-microglobulin (Miyata et al., 1994aSaxena et al., 2000). AGE-modification renders model proteins cytotoxic, whereby the toxicity of a protein increases with its degree of modification (Gasic-Milenkovic et al., 2001). Oxygen free radicals are involved in AGE toxicity because many types of antioxidants, including 17β-estradiol and α-lipoic acid, can block their toxic effects (Loske et al., 1998). However, the mechanisms by which AGEs induce cell death and the accompanying metabolic changes have not yet been elucidated. Thus we have used a neuroblastoma model cell line to investigate the effects of membrane permeable antioxidants on redox-sensitive AGE-induced dysfunctions of glucose metabolism and energy production.
MATERIALS AND METHODS
Production of advanced glycation endproducts
Bovine serum albumin (BSA)-AGE was produced by incubation of 1 mM BSA with 1 M glucose at 50°C in Phosphate-buffered saline (PBS) (pH 7.4) for 6 weeks; the slightly elevated temperature was used to accelerate the reaction and avoid bacterial contamination. Unbound sugars were removed by extensive dialysis with distilled water. All AGE preparations were tested for bacterial contamination using the E-toxate test (Sigma-Aldrich, Taufkirchen, Germany) and were found to be below the detection limit of the endotoxin assay. AGE content was characterized by the optical density at 400 nm and by measuring the specific AGE Carboxymethyllysine (CML) with a commercially available ELISA (Roche Diagnostics) as described previously (Wagner et al., 2001). The concentrations of the AGE-modified proteins were calculated using the molecular weight of the nonglycated protein. Characteristics of the BSA-AGE preparation were as follows: OD400, 0.56/mg protein; and CML, 1.8 nmol/mg protein (0.12 CML residues per molecule of BSA).
Cell culture
Human neuroblastoma cells (SH-SY5Y) were seeded into 24-well plates at a density of 6 × 105 cells/mL. Cells were grown in DMEM supplemented with 10% fetal calf serum, streptomycine/penicilline and 2 mM L-glutamine at 37°C in a humidified atmosphere containing 5% CO2/95% air. After 48 hours, cells were washed with fresh medium, and AGEs and/or antioxidants at the indicated concentrations were added.
Detection of receptor for advanced glycation endproduct(s) by Western Blot
Cells were lysed with lysis buffer containing 0.5% Triton X100 and 1 mM sodium vanadate. A concentration of 21 μg/μL protein from each sample was added to loading buffer, heated 5 minutes at 95°C, and loaded on a 12% SDS polyacrylamide gel. After electrophoresis, proteins were blotted from the gel onto a nitrocellulose membrane by the semi-dry method using a special transfer buffer (250 mM Tris, 20% methanol; 25 mM Tris, 20% methanol; 40 mM 6-amino-n-hexanoic acid, 0.4 M SDS, 20% methanol). After incubation in 10% Rotiblock (Roth, Numberg, Germany) solution for 1 hour at 4°C and repeated washes with TBS/0.05% Tween 20, the membrane was incubated with a polyclonal mouse-anti-receptor for advanced glycation endproduct(s) (RAGE) antibody (1:1000 in TBS/10% Rotiblock) (donated by Dr. B. Weigle, Technical University Dresden) for 1 hour at room temperature. The membrane was washed four times with TBS for 2 minutes and incubated with anti-mouse IgG peroxidase conjugate (1:1000 in TBS/10% rotiblock; DAKO A/S Denmark) for 60 minutes at room temperature. After washing the membrane four times for 2 minutes with TBS/0.05% Tween 20, detection of the marked bands was performed by chemiluminescence (ECL, Amersham).
Determination of ATP concentrations
ATP was measured by an ATP bioluminescence assay kit (HSII, Roche Diagnostics, Germany). Briefly, cells were harvested, resuspended in PBS, and lysed according to the manufacturer's instructions. Samples were immediately frozen at −80°C until analysis with a Berthold one-tube luminometer. ATP concentrations were calculated using a calibration curve of serial ATP dilutions.
Determination of cell numbers
After incubation with Aβ/AGEs or test substances, cells were washed with PBS and trypsinized. Cell culture medium was added, and the single cell suspension was centrifuged for 5 minutes at 800 rpm. Cell numbers were determined with a Schaerfe Casy cell counter equipped with a 150 μm capillary in triplicate experiments. All cells with a size between 9 and 18 μm were considered to be viable.
Determination of lactate concentrations
Lactate concentration in the culture medium was determined in supernatants by the lactate dehydrogenase/glutamate-pyruvate transaminase method. Cell culture medium (20 μL) and distilled water (180 μL) were incubated at 37°C with 200 μL of a reaction mixture containing 250 mM glutamate/NaOH buffer, pH 8.9, 2.8 mM NAD, 0.275 U/mL glutamate-pyruvate transaminase, and 2.75 U/ml lactate dehydrogenase for 60 minutes, after which the absorbance at 340 nm was measured. Lactate concentrations were calculated by comparison with standards in cell culture medium.
Determination of glucose concentrations
Glucose concentration in the culture medium was measured by conversion of glucose to gluconate by glucose-oxidase (Trinder, Sigma). Briefly, 5 μL of the culture supernatants were diluted in 45 μL distilled water and 50 μL of test reagent was added. After 10 minutes of incubation at room temperature, the absorbance at 550 nm was measured. Glucose concentrations were calculated by comparison with standards in cell culture medium.
Statistics
Statistical analysis of the results was carried out by ANOVA with Bonferroni/Dunn adjustment using the SPSS statistics program. Statistical significance was established at ∗ P < 0.05 or ∗∗ P < 0.01, respectively.
RESULTS
Effects of advanced glycation endproducts upon ATP levels
AGEs are structural components of the β-amyloid plaque and have been shown to be cytotoxic by mechanisms involving oxidative stress. Because many types of cell death involve impairment of energy production, we were interested to investigate the effect of AGEs on parameters of glucose metabolism and cellular ATP levels. The human SH-SY5Y neuroblastoma cell line was chosen for this purpose because it is a widely used neuronal cell line for the investigation of neurotoxic compounds (Krishnamurthy et al., 2000; Nath et al., 1997; Zhong et al., 1999), and it constitutively expresses the RAGE in amounts comparable with other cell lines (Fig. 1).
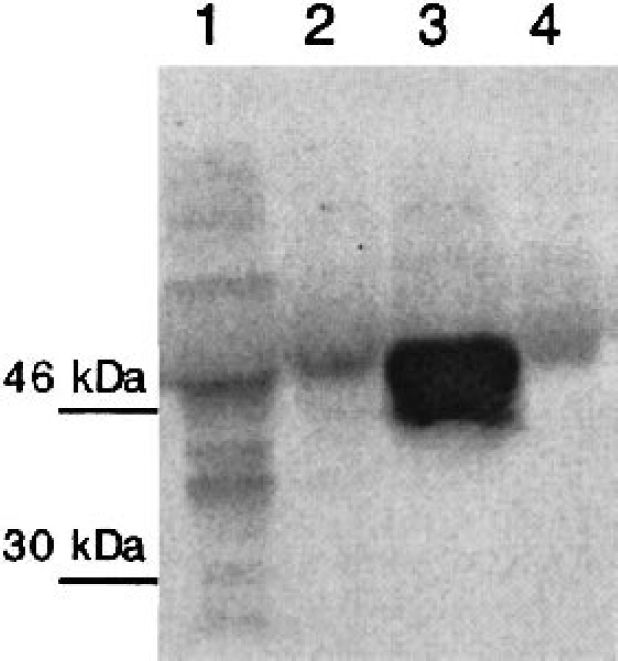
Expression of RAGE in different cell lines. RAGE was detected by Western Blot in SH-SY5Y neuroblastoma cells (lane 1), CaCo-2 human colon carcinoma cells (lane 2), RAGE over-expressing baculovirus infected Sf9 cells (lane 3), and N-11 mouse microglia cells (lane 4). The most abundant RAGE isoform has a molecular weight of approximately 50 kDa. RAGE, receptor for advanced glycation endproduct(s).
In the initial set of experiments, dose- and time-dependent effects of AGEs on ATP concentration were determined. Because it was observed during our experiments that the number of cells (determined by cell counting) decreased upon AGE incubation, all energetic parameters were normalized to cell numbers. Based upon our previous experiments, BSA-AGE (used as a model-AGE) was applied in three different concentrations ranging from 50 to 150 μM for 24 hours, after which the ATP level was determined. AGEs significantly decreased ATP concentration (e.g., to 32% by 150 μM BSA-AGE and to 58% by 100 μM BSA-AGE after 24 hours) in a time- and dose-dependent manner (Fig. 2). Nonmodified BSA had no significant effect on ATP levels.
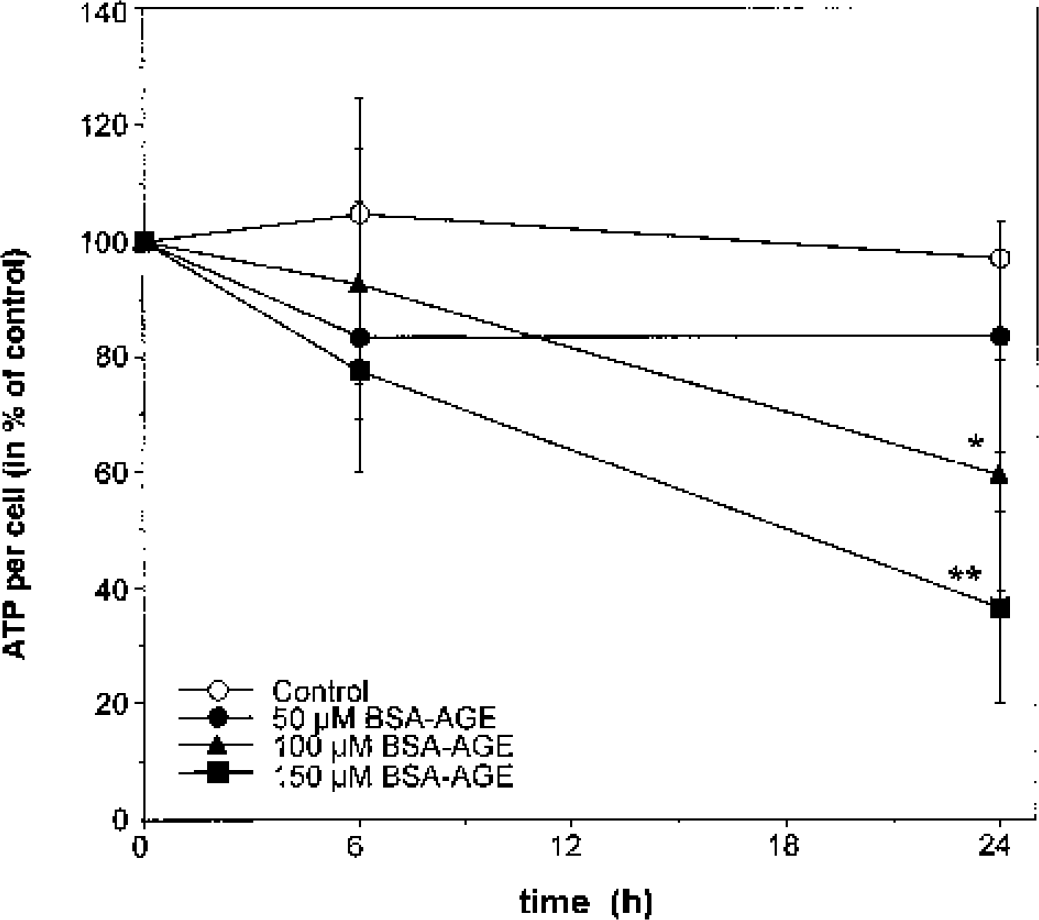
Time-dependent changes in ATP level of SH-SY5Y neuroblastoma cells by AGEs. BSA-AGE decreases ATP level per cell of SH-SY5Y neuroblastoma cells in a dose- and time-dependent manner. Cells were incubated with 50 μM, 100 μM, or 150 μM BSA-AGE for up to 24 hours. ATP content of cells was measured after 0, 6, and 24 hours. 100% was defined as ATP per cell at t = 0. A significant decrease of the intracellular ATP content was detected at concentrations of 100 and 150 μM BSA-AGE compared with untreated cells after 24 hours. Data are taken from three separate experiments (each done in duplicate) and are expressed as mean ± SD. ∗ P < 0.05; ∗∗ P < 0.01. AGE(s), advanced glycation endproduct(s); BSA, bovine serum albumin.
AGE mediated cytotoxicity was previously shown to be mediated by oxygen free radicals (Loske et al., 1998). Because a variety of membrane permeable antioxidants, including 17β-estradiol and α-lipoic acid, could protect neuronal and nonneuronal cells from AGE-induced cell death (Loske et al., 1998), we also investigated their effects on the AGE-induced changes in energy metabolism. 17β-estradiol is an effective phenolic antioxidant in aqueous phase with predominant superoxide radical scavengeing ability (Ayres et al., 1998), and α-lipoic acid (or more accurately dihydrolipoate, its intracellularly reduced form) is a versatile scavenger. It reacts with reactive oxygen species, predominantly with hydroxyl radicals, hypochlorous acid, peroxyl radicals, singlet oxygen, and nitric oxide (Packer et al., 1995). The antioxidant concentrations (100 μM (R+)-α-lipoic acid/10 μM 17β-estradiol) were taken from previous cytotoxicity experiments, where they protected 80% to 85% of cells from AGE-induced cell death (Loske et al., 1998). In addition, aminoguanidine (carbonyl scavenger, antioxidant, and NO synthase inhibitor), pyruvate (energy substrate and antioxidant), and catalase were also tested for their protective effects on ATP levels upon exposure of cells to AGEs. When administered 1 hour before the AGEs, all the antioxidants were able to restore (at least partially) the AGE-induced decrease in ATP level in cells stressed with 100 μM BSA-AGE (Fig. 3), suggesting that the AGE induced ATP depletion involves the production of ROS such as superoxide or its dismutation product, hydrogen peroxide.
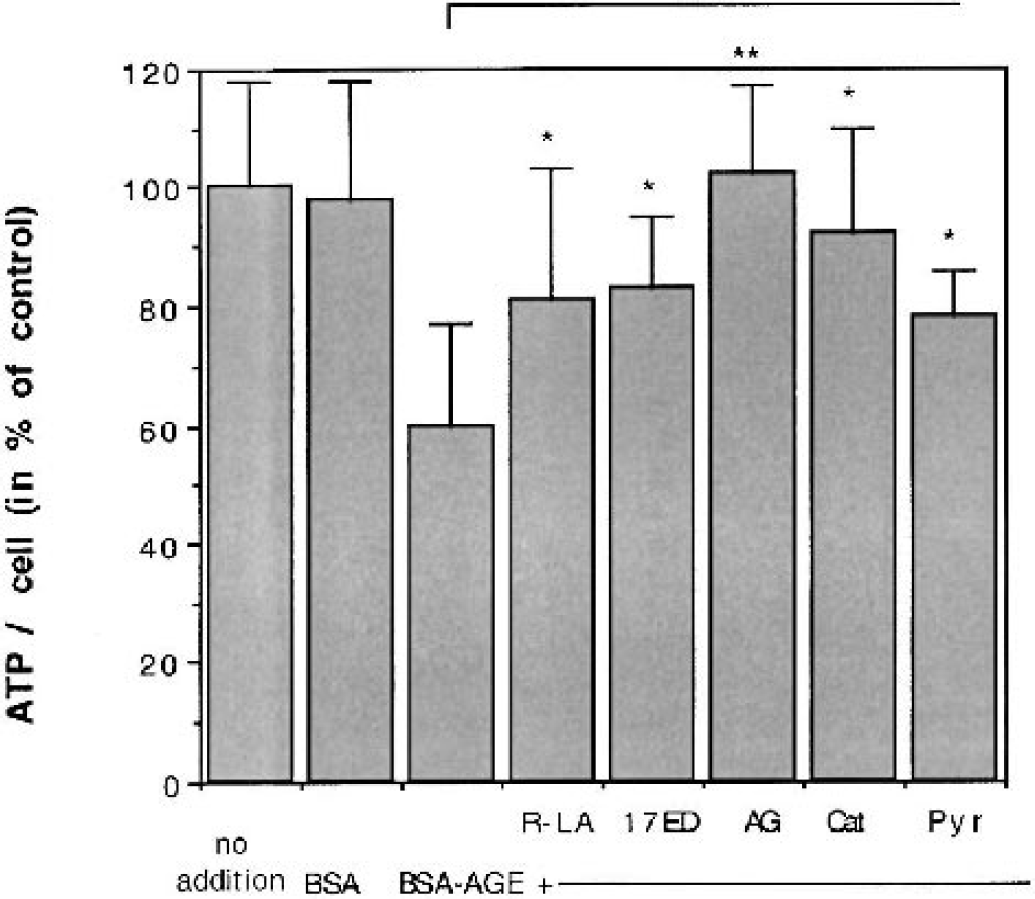
Effects of antioxidants upon ATP levels in SH-SY5Y neuroblastoma cells treated with AGEs. ATP levels of AGE-treated cells are significantly increased after treatment with 100 μM (R+)-α-lipoic acid (LA), 10 μM 17β-estradiol (17ED), 2 mM aminoguanidine (AG), 400 μg/mL catalase (Cat), and 2 mM pyruvate (Pyr), compared with cells treated with BSA-AGE only. Cells were preincubated for 1 hour with the antioxidants before 100 μM BSA-AGE was added. The ATP content per cell was determined after 24 hours. Data are taken from four separate experiments (each done in duplicate) and are expressed as mean ± SD. ∗ P < 0.05; ∗∗ P < 0.01. AGE(s), advanced glycation endproduct(s); BSA, bovine serum albumin.
Effects of advanced glycation endproducts upon glucose consumption and lactate production
AGE-induced decrease in ATP concentration points to an impaired glucose flux, which could be caused by an inhibition of the activity of enzymes of the glycolytic pathway, of the Krebs cycle, or of mitochondrial function. To investigate more details of AGE-induced changes in glucose metabolism, we measured their effects upon glucose consumption (measured as uptake from the cell culture medium) and lactate production (measured as lactate release into the medium). Glucose consumption was determined indirectly by measuring the glucose remaining in the culture supernatant after incubation with AGEs. No increase in glucose consumption compared with control was detected in cells incubated with 50 μM BSA-AGE. At 100 μM BSA-AGE, glucose consumption increased up to fivefold during a 24-hour period (Fig. 4 and Fig. 5). When glucose consumption was followed over time, it appeared that the highest rate of glucose uptake from the medium occurred during the first 2 to 6 hours after AGE exposure (Fig. 4). Lactate production was increased already at a concentration of 50 μM BSA-AGE, reaching a maximum (greater than fivefold increase) in cells incubated with 100 μM BSA-AGE (Fig. 5). Similar to the results observed with ATP, glucose consumption and lactate production were normalized by all the antioxidants tested, as well as by the glycolytic metabolite pyruvate (Fig. 5). These data indicate that the AGE-induced disturbances in glucose metabolism and the resulting shift to anerobic glycolysis are mediated by oxygen free radicals.
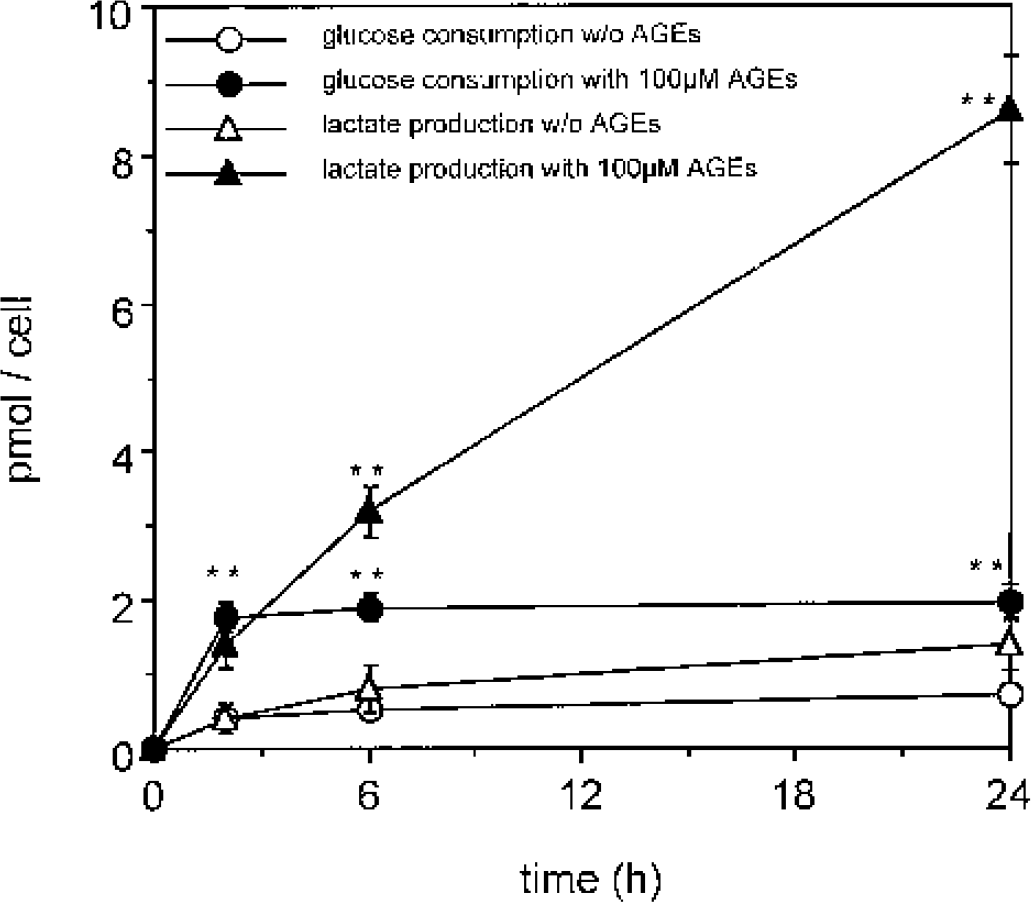
Time-dependent changes in glucose consumption and lactate production by AGEs. Cells were incubated with 100 μM BSA-AGE. Glucose and lactate concentrations in the medium were determined after 2, 6, and 24 hours, and the molar amount of used glucose consumption and lactate production was calculated per cell. A significant difference of glucose consumption and lactate production could be observed in samples incubated with AGEs (filled symbols) compared with those without AGEs (empty symbols) after 2, 6, and 24 hours. Data are taken from four separate experiments and are expressed as mean ± SD. ∗∗ P < 0.01 compared with the control (without AGE at the same time point). AGE(s), advanced glycation endproduct(s); BSA, bovine serum albumin.
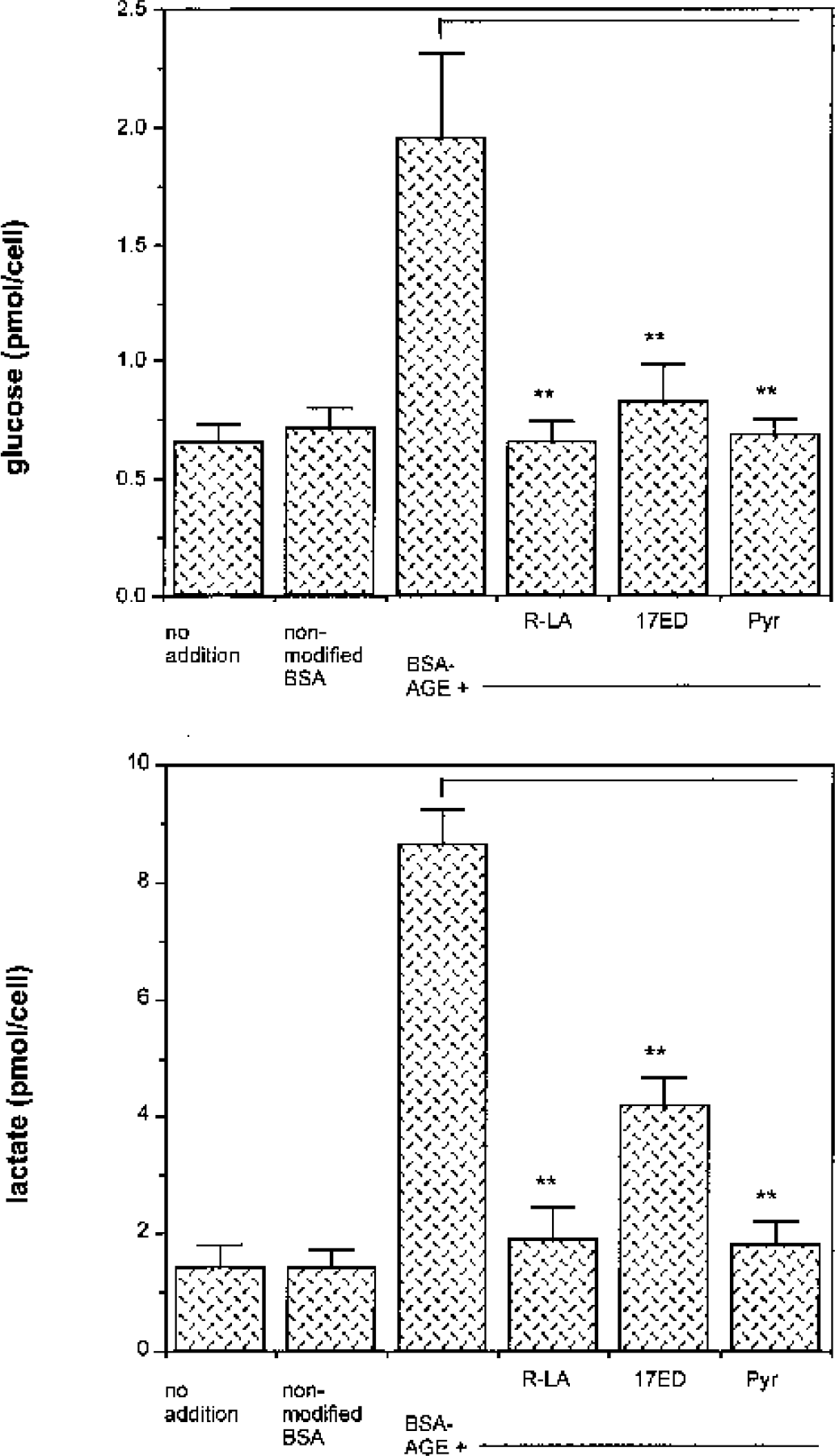
Effect of antioxidants on glucose consumption and lactate production in cells treated with BSA-AGE. AGE-induced increase in glucose consumption and lactate production is attenuated by membrane permeable antioxidants. Cells were preincubated with 100 μM (R+)-α-lipoic-acid (LS), 10 μM 17β-estradiol (17ED), or 2mM pyruvate (Pyr) for 1 hour before administration of 100 μM BSA-AGE for 24 hours. A significant difference between samples treated with BSA-AGE only and the samples with added antioxidants was observed for both glucose consumption and lactate production after 24 hours. Data are taken from four separate experiments (each done in duplicate) and are expressed as mean ± SD. ∗∗ P < 0.01. AGE(s), advanced glycation endproduct(s); BSA, bovine serum albumin.
DISCUSSION
AGEs accumulate upon β-amyloid plaques of AD and are suggested to contribute to oxidative stress, cell damage, functional loss, and even neuronal cell death in the AD brain. However, chemically defined AGEs, such as CML or pentosidine, have not been quantified in senile plaques so far. However, plaque fractions from AD brains analyzed by a standardized ELISA were found to contain approximately threefold more AGE adducts than preparations from healthy, age-matched controls and in the same order of magnitude than synthetically prepared AGEs (Amyloid-AGE) incubated in 1 M glucose for 4 months (Vitek et al., 1994). If one considers an amyloid plaque as a compact protein deposit, the local concentration of AGEs such as CML could be estimated to be in the low millimolar range. In summary, plaque proteins are likely to be as extensively modified by AGE as other long-lived proteins, including eye lens crystallin and β2-microglobulin (Miyata et al., 1994a; Saxena et al., 2000).
In this study, we show that AGEs lead to increase in lactate production and to a decrease in ATP levels. In addition, glucose consumption is increased in the early phase of AGE exposure. These data suggest that AGEs impair glucose flux through the Krebs cycle and oxidative phosphorylation, resulting in the conversion of pyruvate to lactate rather than to acetyl-CoA. Of interest is the fact that glucose consumption is inhibited at later time points, which could be explained by different mechanisms: it could be caused (a) by the loss of glucose transport systems, such as an inactivation of glucose transporters (Keller et al., 1997), (b) by an inactivation of pyruvate dehydrogenase by its allosteric effector NADH, or (c) by inhibition of glycolytic or mitochondrial enzymes (e.g., pyruvate dehydrogenase, aconitase, or α-ketoglutarate dehydrogenase) by AGE-induced lipid peroxidation products (Humphries and Szweda, 1998). However, the high demand of glucose could also be driven by the necessity to produce reductive equivalents (NADPH) via pentose phosphate shunt for antioxidative defenses, such as regeneration of glutathione.
Many of the AGE-induced changes in glucose metabolism have also been reported to be present in diabetes, including increased plasma lactate (Thurston et al., 1995) or low ATP levels in cells of diabetic mice (Liu et al., 1999) and to bear close resemblance to conditions occurring in hypoxia. In diabetes, these metabolic disturbances are referred to as metabolic pseudohypoxia because tissue oxygen concentration is normal (Williamson et al., 1993). Signal transduction of AGEs is transmitted RAGE, which is expressed in a variety of cells and tissues (Fig. 1). RAGE is a multiligand member of the immunoglobulin superfamily of cell surface molecules whose repertoire of ligands includes advanced glycation end products (AGEs), amyloid fibrils, amphoterins, and S100/calgranulins. AGE ligands tethered to the complex of RAGE and lactoferrin-like polypeptide results in induction of oxidant stress, including the generation of thiobarbituric acid reactive substances and activation of NF-κB, each of which is blockable by antibodies to RAGE receptor polypeptides and by antioxidants (Yan et al., 1994). AGEs also behave as active ligands for some scavenger receptors, including class A scavenger receptor and class B scavenger receptors such as CD36 and scavenger receptor, class B, type I (Miyazaki et al., 2002).
Of interest is the fact that many of the AGE-induced metabolic changes have been reported in the AD brain, too. For example, increased levels of lactic acid are present in the cerebrospinal fluid (CSF) of AD patients (Parnetti et al., 1995; Redjems-Bennani et al., 1998).
Mechanistically, the involvement of reactive oxygen and nitrogen species in AGE-induced changes in metabolic functions is supported by the efficacy of the membrane permeable antioxidants such as (R+)-α-lipoic acid and 17β-estradiol. 17β-estradiol has predominant superoxide radical scavenging abilities (Behl and Moosmann, 2002), whereas (R+)-α-lipoic acid reacts with superoxide, hydroxyl radicals, hypochlorus acid, peroxyl radicals, singlet oxygen, and nitric oxide (Packer et al., 1995). (R+)-α-lipoic can scavenge α-oxoaldehydes and lipid peroxidation products very efficiently (Nickander et al., 1996). Second, it can regenerate glutathione directly, which is important for oxoaldehyde detoxification and regeneration of other antioxidants like vitamin E via redox cycling (Packer et al., 1995). (R+)-α-lipoic acid was particularly effective in reducing lactate production to control values, an effect also observed in clinical trials with diabetic patients (Konrad et al., 1999). (R+)-α-lipoic acid-supplemented old rats have also been shown to have improved mitochondrial function, decreased oxidative damage, and increased metabolic rate (Hagen et al., 1999). The concentrations of antioxidants used in this study to determine the mechanisms of AGE-induced changes in metabolism are slightly higher than the therapeutically achievable concentrations. For example, when 600 mg α-lipoic acid was given as a single oral infusion to patients with type II diabetes, plasma levels of up to 32 μM could be measured (Rosak et al, 1996). Although α-lipoic acid concentrations have not been determined in human brain tissue, some data on plasma and tissue levels of therapeutically administered α-lipoic acid are available from animal studies. When a single dose of 10 mg/kg lipoic acid was given per os (PO) or intravenously to rats, the maximal concentration of lipoic acid in plasma reached approximately 160 μg/mL (7.8 μM). When α-lipoic acid was fed for 21 consecutive days, lipoic acid was found to accumulate in peripheral nerves (N. ischiaticus and N. femoralis) at a concentration 10 times higher than that of plasma when measured after 24 hours (Peter et al, 1996). However, this study used radioactively labeled α-lipoic acid, and the authors calculated that approximately 50% of the radioactivity could be already converted into other metabolites, such as by β-oxidation of the fatty acid moieties (Peter et al, 1996). 17β-estradiol at a concentration of 10 μM cannot be used in clinical practice because of its hormonal action. However, scavestrogens, 8,9-dehydro derivatives of 17α-estradiol and 17β-estradiol, have improved radical scavenging activity and neglectable estrogen receptor activation and are superior to 17β-estradiol as neuroprotective agents (Römer et al, 1997).
Superoxide was suggested to be a mediator in AGE-induced metabolic disturbances (Ortwerth et al., 1998; Yan et al., 1995), most likely produced by activation of NADPH oxidase (Wautier et al., 2001). Most likely, AGEs may lead to ROS-induced inactivation of key respiratory chain complexes such as complex I, II-III, and IV, as well as the tricarboxylic acid cycle enzyme aconitase (Melov et al., 1999) and the creatine kinase enzyme system by oxgen free radicals (Fig. 6).
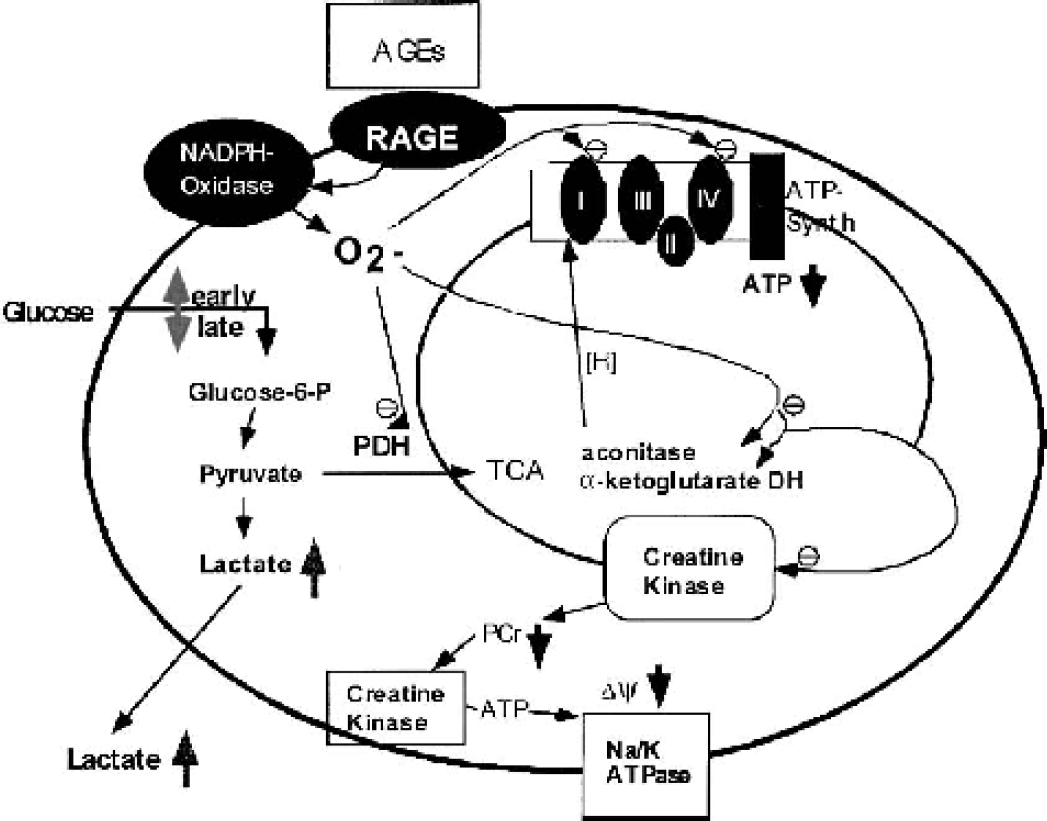
Hypothetical model of AGE-induced radical production and subsequent impairment of energy production. Binding of AGEs to RAGE results in increased production of ROS, which inactivates ROS-sensitive enzymes including those of the TCA cycle and the mitochondrial respiratory chain. This could lead to the impairment of glucose flux through pyruvate dehydrogenase and/or the mitochondrial respiratory chain and subsequently to decreased ATP levels (with consequences for the neuronal membrane potential) and increased lactate production. AGE(s), advanced glycation endproduct(s); RAGE, receptor for advanced glycation endproduct(s).
In summary, we believe that AGEs, among other neurotoxic factors such as β-amyloid peptide, inflammation, and other (yet unknown) processes, can contribute to disturbances in glucose metabolism and to impaired energy production in neurons in the AD brain. Thiol antioxidants such as (R+)-α-lipoic acid and nonhormonal derivatives of 17β-estradiol may become promising therapeutic drugs for the treatment of AGE-mediated cellular energy depletion, which is suggested as one of the underlying causes of AD (Adair et al., 2001; Hager et al., 2001).
Footnotes
Abbreviations used:
Acknowledgment
We thank P. Riederer, Th. Arendt, D. Palm, M. Götz, K. Hager, M. Kenklies, and M. Gerlach for helpful and stimulating discussions.