
Abstract
The role of brain insulin-like growth factors (IGFs) and IGF binding proteins (IGFBPs) in neuroprotection was further investigated using in vitro and in vivo models of cerebral ischemia by assessing the effects of IGF-I, IGF-II, and high affinity IGFBP ligand inhibitors (the peptide [Leu24, 59, 60, Ala31]hIGF-I (IGFBP-LI) and the small molecule NBI-31772 (1-(3,4-dihydroxybenzoyl)-3-hydroxycarbonyl-6, 7-dihydroxyisoquinoline), which pharmacologically displace and elevate endogenous, bioactive IGFs from IGFBPs. Treatment with IGF-I, IGF-II, or IGFBP-LI (2 μg/mL) significantly (P < 0.05) reduced CA1 damage in organotypic hippocampal cultures resulting from 35 minutes of oxygen and glucose deprivation by 71%, 60%, and 40%, respectively. In the subtemporal middle cerebral artery occlusion (MCAO) model of focal ischemia, intracerebroventricular (icv) administration of IGF-I and IGF-II at the time of artery occlusion reduced ischemic brain damage in a dose-dependent manner, with maximum reductions in total infarct size of 37% (P < 0.01) and 38% (P < 0.01), respectively. In this model of MCAO, icv administration of NBI-31772 at the time of ischemia onset also dose-dependently reduced infarct size, and the highest dose (100 μg) significantly reduced both total (by 40%, P < 0.01) and cortical (by 43%, P < 0.05) infarct volume. In the intraluminal suture MCAO model, administration of NBI-31772 (50 μg icv) at the time of artery occlusion reduced both cortical infarct volume (by 40%, P < 0.01) and brain swelling (by 24%, P < 0.05), and it was still effective when treatment was delayed up to 3 hours after the induction of ischemia. These results further define the neuroprotective properties of IGFs and IGFBP ligand inhibitors in experimental models of cerebral ischemia.
Insulin-like growth factors I and II (IGF-I and IGF-II) are multifunctional polypeptides, which are essential for normal growth and development (Jones and Clemmons, 1995; Stewart and Rotwein, 1996). Alterations in the IGF system are implicated in a variety of pathologic conditions, including brain injury. The expression of IGF in the brain is increased after ischemic, hypoxic, or physical brain injury (Beilharz et al., 1995; Lee et al., 1996; Sandberg Nordqvist et al., 1996; Breese et al., 1996; Walter et al., 1997), and administration of exogenous IGF reduces brain damage and improves functional outcome after hypoxic-ischemic (Guan et al., 1993; Guan et al., 1996; Johnston et al., 1996; Guan et al., 2000c; Guan et al., 2001) and focal ischemic (Loddick et al., 1998; Wang et al., 2000; Schabitz et al., 2001; Liu et al., 2001a) brain injuries in rodents.
Most biologic actions of IGFs appear to be mediated by the type-I IGF receptor (Ullrich et al., 1986; Czech, 1989) and modulated by the IGF-binding proteins (IGFBPs). In body fluids, including cerebrospinal fluid (CSF), the majority of IGFs are associated with IGFBPs, which bind IGFs with high affinity and have an inhibitory effect by limiting their interaction with the receptor (Clemmons, 1998; Schneider et al., 2000; Silha and Murphy, 2002). Although some IGFBPs have been proposed to facilitate IGF actions (Guan et al., 2000a), a potential reserve of IGF appears to exist, which if released from the IGFBPs would elevate the levels of free biologically active IGF. The authors hypothesized and recently demonstrated that this reserve of bound IGF provides a unique opportunity to exploit an endogenous neuroprotective agent. We used an analog of IGF, which has no biologic activity at the IGF receptors but binds to the IGFBPs and elevates the levels of biologically active IGF in the CSF (Loddick et al., 1998). We demonstrated that intracerebroventricular (icv) administration of this analog, [Leu24, 59, 60, Ala31]hIGF-I (IGF binding protein ligand inhibitor, IGFBP-LI) mimicked the effects of IGF-I and dramatically reduced brain damage after experimental cerebral ischemia in the rat (Loddick et al., 1998). A similar analog, which also possesses the ability to displace IGF from IGFBPs, has been shown to mimic the effects of IGF-I in animal models of diabetes and growth failure (Lowman et al., 1998). Thus the use of such an analog appears to be an attractive strategy for elevating levels of biologically active IGF. However, clinical applications for a peptide-based therapy are limited, and such a strategy would also elevate the levels of free biologically active IGF-II, which is less studied than IGF-I. IGF-II is present in the injured brain (Beilharz et al., 1995; Walter et al., 1999), but its role is unclear. Whereas protective effects of IGF-II have been demonstrated in vitro (Cheng and Mattson, 1992; Nicholas et al., 2002), after experimental hypoxic ischemic brain injury in vivo, IGF-II failed to reduce damage, and coadministration of IGF-II reduced the neuroprotective effects of IGF-I (Guan et al., 1996). Such a detrimental effect of IGF-II would limit the potential benefits of a therapeutic strategy that releases endogenous IGFs into the brain.
The present study sought to further explore the role of brain IGFs and IGFBPs in neuroprotection in experimental ischemia both in vitro and in vivo. The neuroprotective effects of IGF-I, IGF-II, and IGFBP-LI were investigated in vitro in the organotypic hippocampal culture model of ischemia. Organotypic hippocampal cultures provide a method of determining direct neuronal effects of a drug on ischemic injury in the absence of, for example, the circulation and infiltrating inflammatory cells. In vivo, the effects of the IGF peptides were determined in two rat models of permanent MCA occlusion, and the aims were threefold. First, the dose-response effects of IGF-1 and IGF-II on focal ischemic damage were assessed when administered immediately after MCA occlusion. Second, the dose-effect relationship of NBI-31772 (1-(3,4-dihydroxybenzoyl)-3-hydroxycarbonyl-6, 7-dihydroxyisoquinoline) on infarction was investigated when administered immediately after the onset of ischemia. NBI-31772 is a recently discovered nonpeptide small molecule that binds to all six known IGFBPs and displaces biologically active IGF-I (Chen et al., 2001; Liu et al., 2001b). Third, the extent of the therapeutic time window for a neuroprotective dose of NBI-31772 in permanent focal ischemia in the rat was determined.
MATERIALS AND METHODS
All experiments were conducted in accordance with the United Kingdom Animals (Scientific Procedures) Act (1986) or were approved by the Institutional Animal Care and Use Committee of Neurocrine Biosciences in accordance with National Institutes of Health Guidelines. Human recombinant IGF-I and IGF-II were purchased from Austral Biologicals (San Ramon, CA, U.S.A.). The Departments of Medicinal and Peptide Chemistry synthesized IGFBP-LI and NBI-31772, as previously described (Loddick et al., 1998; Chen et al., 2001).
Organotypic hippocampal slice culture
Culture preparation
Organotypic hippocampal slice cultures were prepared as described previously (Stoppini et al., 1991) with modifications. Sprague Dawley rat pups, 6 to 8 days old (Charles River Laboratories, Raleigh, NC, U.S.A.), were killed by decapitation, and the hippocampi were rapidly dissected out. Using a McIlwain tissue chopper, 400 μm thick sections were cut and placed into ice-cold growth medium. The growth medium consisted of 50% minimum essential medium (MEM), 25% Hank's balanced salt solution (HBSS), 25% heat-inactivated horse serum, supplemented with 5 mg/mL glucose, 1 mM glutamine, and 1.5% fungizone. Cultures were placed onto semiporous membranes and then grown for 10 to 14 days in an incubator at 37°C with 5% CO2 enriched atmosphere before being viable for use in reverse transcription-polymerase chain reaction (RT-PCR) analysis and oxygen-glucose deprivation (OGD) (i.e., in vitro ischemia) experiments.
RT-PCR analysis
Total RNA was isolated from cultured organotypic hippocampi at 14 days using Trizol (Life Technologies, Carlsbad, CA, U.S.A.). From this RNA, cDNA was generated by first strand synthesis using Ambion's RETROscript kit and first strand random decamers. cDNAs of interest were then used in PCR reactions with the 10 sets of primers for the genes of interest. The individual products of interest were analyzed by size separating products using agarose gel electrophoresis and visualizing bands with ethidium bromide, as described previously (Naeve et al, 2000). Proper function of the primer pairs that did not amplify with slice culture cDNA was verified using cDNA prepared from brain regions or tissue in which the appropriate binding protein is known to be expressed.
Oxygen-glucose deprivation
Organotypic hippocampal cultures were placed in serum-free medium containing 75% MEM, 25% HBSS, 1 mM glutamine, and 1.5% fungizone 1 hour pre-OGD. The cultures were then transferred to six-well plates containing glucose-free medium (75% glucose-free MEM, 25% HBSS, 1 mM glutamine, and 1.5% fungizone) saturated with 95% N2, 5% CO2 and sealed into a custom built, plexiglass anaerobic chamber equipped with inlet and outlet valves that was equilibrated to 37°C, 100% humidity. The 95% N2, 5% CO2 gas mixture was blown through the chamber for 10 minutes before the chamber was sealed for a 35 minute period of OGD. On removal of the plates from the chamber at the end of the OGD, the inserts were transferred to prewarmed serum-free medium containing 5 μg/mL propidium iodide (PI) and then placed back into the CO2 incubator for 48 hours. IGF-I, IGF-II, and IGFBP-LI were dissolved in distilled water at a concentration of 2 μg/mL, and they were added to the medium immediately before exposure to OGD and subsequently maintained in the growth medium for 48 hours.
Analysis of damaged CA1 hippocampal neurons after OGD was carried out using ImagePro Plus Software (Media Cybernetics, Silver Spring, MD, U.S.A.) as described by Newell and co-workers (1995). Briefly, the intensity of PI was used as an index of cell death. The first measurement of fluorescent intensity was performed at 48 hours after initial OGD. The remaining neurons were killed by exposing the cultures to 3 hours of anoxia. The fluorescent intensity obtained 24 hours after 3 hours of anoxia was set equal to 100% damage and was then compared with the fluorescent intensity at 48 hours after the initial OGD. The integrated fluorescent intensity from the CA1 after the initial insult was then expressed as a percentage of the value obtained after 3 hours of anoxia (complete damage).
Focal cerebral ischemia
Surgical preparation
Male Sprague-Dawley rats, weighing 160 to 190 g, were anesthetized with isoflurane (4% induction, 2% maintenance) in 100% O2, and indwelling guide cannulae were stereotaxically implanted in the right, lateral ventricle (coordinates in mm: lateral [+1.5]; anteroposterior [−0.8]; dorsoventral [−3.0] relative to Bregma) to permit subsequent injections of peptides or NBI-31772 into the CSF. All rats were allowed to recover for a least 7 days before they were anesthetized initially with a mixture of 5% halothane, 30% oxygen, and 70% nitrous oxide in an induction chamber and maintained thereafter with a nitrous oxide/oxygen mixture (70%: 30%) containing 1.0% to 1.5% halothane delivered via a face mask for induction of permanent focal cerebral ischemia. Body temperature was monitored throughout all surgical procedures by a rectal thermometer, and the animals were maintained normothermic (37 ± 1°C) via a heating blanket controlled by the thermometer.
Subtemporal middle cerebral artery occlusion model
Permanent focal cerebral ischemia was induced in male Sprague-Dawley rats (240–280 g; Charles River, U.K.) by electrocoagulation of the left middle cerebral artery (MCA) using a subtemporal approach, as described previously (Tamura et al., 1981). Briefly, a craniotomy was performed, the dura was opened, and the artery was exposed and occluded from its origin to the point where it crossed the inferior cerebral vein. The artery was then transected to confirm complete occlusion and to prevent recanalization. The incision sites were sutured closed, and the animals were allowed to recover from anesthesia.
The neuroprotective effects of IGF-I, IGF-II, and NBI-31772 were examined in three separate studies after permanent focal ischemia using the subtemporal approach. In each study, a range of doses was tested against the vehicle (5 μL water), and all dosing solutions were adjusted to pH 7 for injection. Vehicle or drug was injected into the lateral ventricle over 2 to 3 minutes immediately after middle cerebral artery occlusion (MCAO), and damage was assessed 24 hours later. No enlargement of the ventricles was observed in any animal when the brains were processed for histologic analysis of infarct volume. Doses for IGF-I and IGF-II were chosen based upon the authors' previous study (Loddick et al., 1998). The doses of NBI-31772 were based upon the authors' unpublished observation that 60 μg of NBI-31772 (administered icv immediately after MCAO) caused a significant reduction of the total lesion volume after permanent MCAO compared with vehicle (vehicle: 133 ± 39 mm3, n = 6 vs. NBI-31772: 65 ± 19 mm3, n = 7, P < 0.0001, data not shown).
Animals were killed 24 hours after MCAO, their brains were removed, and 1mm fresh coronal sections were cut on a vibratome. Brain sections (approximately 12 per brain) were incubated in a 2% solution of triphenyl tetrazolium chloride (TTC), which stains for viable tissue, at 37°C for 30 minutes. Areas of infarct on each brain section were transcribed onto scale diagrams and quantified by computer based image analysis. The volume of infarcted tissue in the hemisphere, cortex and striatum was computed by summing the areas of damage from each section and multiplying them by the distance between the sections.
Intraluminal suture middle cerebral artery occlusion model
The left MCA was occluded permanently in male Sprague-Dawley rats (280–340 g; Charles River, U.S.A.) through a cervical carotid approach using the intraluminal suture technique described previously (Longa et al., 1989). Briefly, the common carotid, external carotid, and internal carotid arteries were exposed through a midline cervical incision. A 30 mm length of poly-l-lysine coated 3–0 monofilament nylon suture (with a heat-blunted tip) (Belayev et al., 1996) was inserted into the external carotid artery and advanced into the lumen of the internal carotid artery until mild resistance was felt (19–20 mm, depending upon body weight), thereby occluding the origin of the MCA. After closure of the skin incision, rats were allowed to recover from anesthesia.
The time window for the neuroprotective effect of NBI-31772 was examined in this model of permanent focal ischemia. A neuroprotective dose of NBI-31772 (50 μg; pH 7) or vehicle (water, 5 μL) was injected icv over 2 to 3 minutes (via previously implanted guide cannula) at 0, 1, 2, or 3 hours after MCAO; ischemic damage was assessed 24 hours later.
Twenty-four hours after MCAO, the animals were killed, and the brains were removed and frozen in isopentane at −40°C. Brains were cut into 20 μm serial coronal sections at (1 mm apart, covering the entire forebrain), which were fixed for 20 minutes in 4% paraformaldehyde and rinsed and stained with hematoxylin and eosin. The sections were examined by light microscopy, and areas of ischemic damage were delineated and transcribed onto scale diagrams. Areas of damage were quantified using a computer-based image analysis system, and the total volumes of ischemic tissue for each brain were derived from integration of the areas of damage in the 12 planes and the known stereotactic coordinates of the planes. The volumes of the cerebral hemispheres ipsilateral and contralateral to the occluded MCA were determined directly from the stained histologic sections by assessment of the total surface area at the same coronal planes as used for assessing areas of ischemic damage. The difference between the two hemispheres provided a measure of brain swelling.
Statistical analysis
All data are expressed as mean ± SD. Significant differences in volume of ischemic damage and percentage of CA1 neuronal damage between vehicle-treated control and drug treatment groups were made using analysis of variance (ANOVA) followed by Dunnett's post hoc test. The relationship between the volume of brain swelling and volume of infarction was assessed by linear regression analysis (Pearson's). For all statistical analyses, the significance level was accepted to be P < 0.05.
RESULTS
Organotypic slice cultures
To characterize the components of the IGF system that are present, an RT-PCR analysis of IGF family mRNA prepared from 14 day old cultured organotypic hippocampal slices was undertaken (Fig. 1). The peptides IGF-I and IGF-II and their associated receptors IGF-RI and IGF-RII were expressed in the hippocampal slice cultures (Fig. 1). The expression profile of six IGFBPs was also determined in the organotypic slice culture. IGF-BP3, IGF-BP5, and IGF-BP6 were expressed in the hippocampus, whereas there was no detectable expression of IGF-BP1, IGF-BP2, or IGF-BP4 (Fig. 1).

RT-PCR analysis of IGF family mRNA from cultured organotypic hippocampus. Total RNA was isolated from slices grown for 2 weeks in vitro and subject to first strand cDNA synthesis. Aliquots of cDNA were added to PCR reactions containing each of the indicated primer sets. Individual PCR products were analyzed by size separating products using agarose gel electrophoresis and visualizing bands with ethidium bromide. RT-PCR, reverse transcription-polymerase chain reaction.
Marked PI fluorescence was observed in the organotypic hippocampal cultures when measured 48 hours after 35 minutes of OGD (Fig. 2). The fluorescence was predominantly located in the CA1 subfield of the hippocampus (Fig. 2). Administration of the IGF peptides and IGFBP-LI significantly reduced hippocampal CA1 cell death produced by OGD (Fig. 2). At a bath concentration of 2 μg/mL, IGF-I, IGF-II, and IGFBP-LI significantly reduced cell death by 71%, 60%, and 40% respectively (P < 0.05).
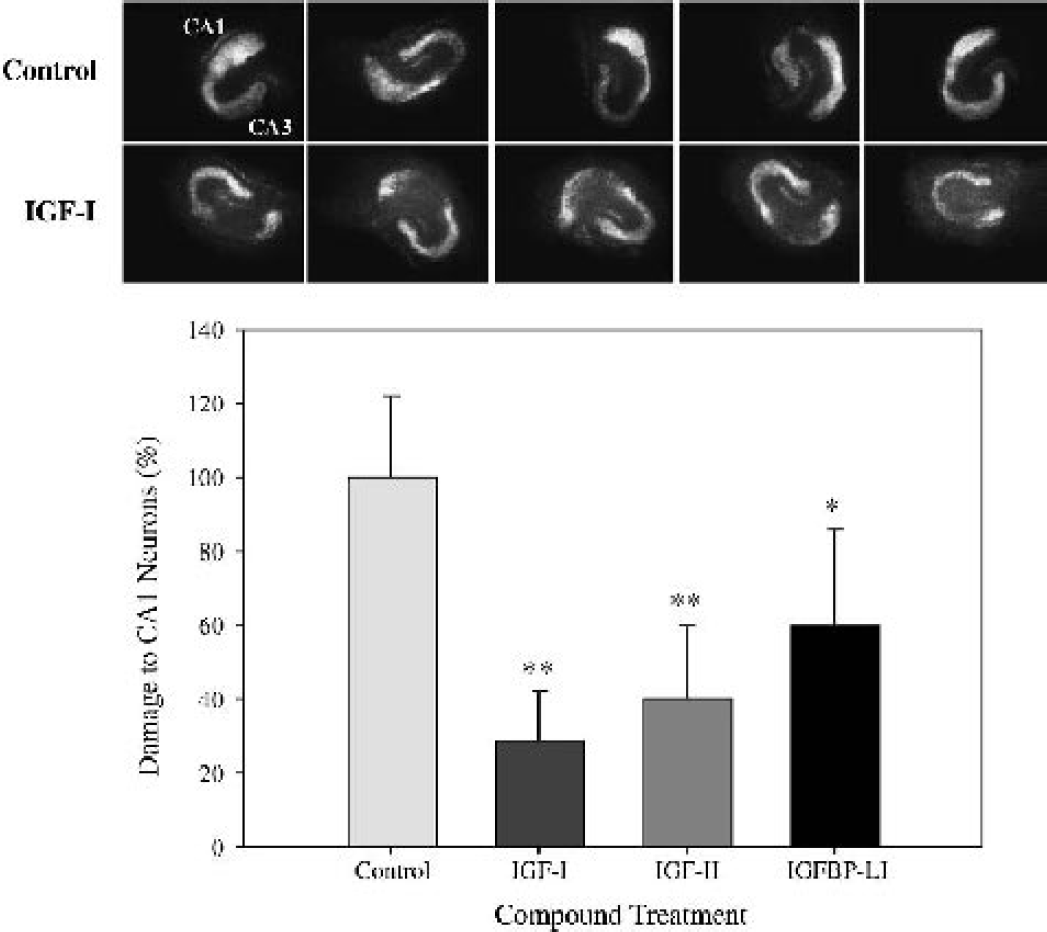
Neuroprotective effects of CA1 pyramidal neurons by IGF-I, IGF-II and IGFBP-LI on OGD in organotypic hippocampal brain slices.
Focal ischemia
Permanent occlusion of the left MCA via the subtemporal approach resulted in ischemic damage only within the territory of the occluded MCA, that is, the frontal, sensory motor and parietal cortices, and the striatum. The effect of IGF-I on ischemic brain damage after permanent focal cerebral ischemia (subtemporal approach) was dose dependent (Fig. 3). Compared with vehicle, the lowest dose tested (5 μg) caused a significant reduction of the striatal lesion volume (30%, P < 0.01). The reduction of the cortical (7%) and total (11%) lesion volume was small and not statistically significant. The two higher doses (25 and 50 μg) caused a statistically significant reduction of the total (32% and 37%, respectively), cortical (30% and 34%, respectively), and striatal (39% and 54%, respectively) lesion volume, compared with vehicle treatment (Fig. 3).
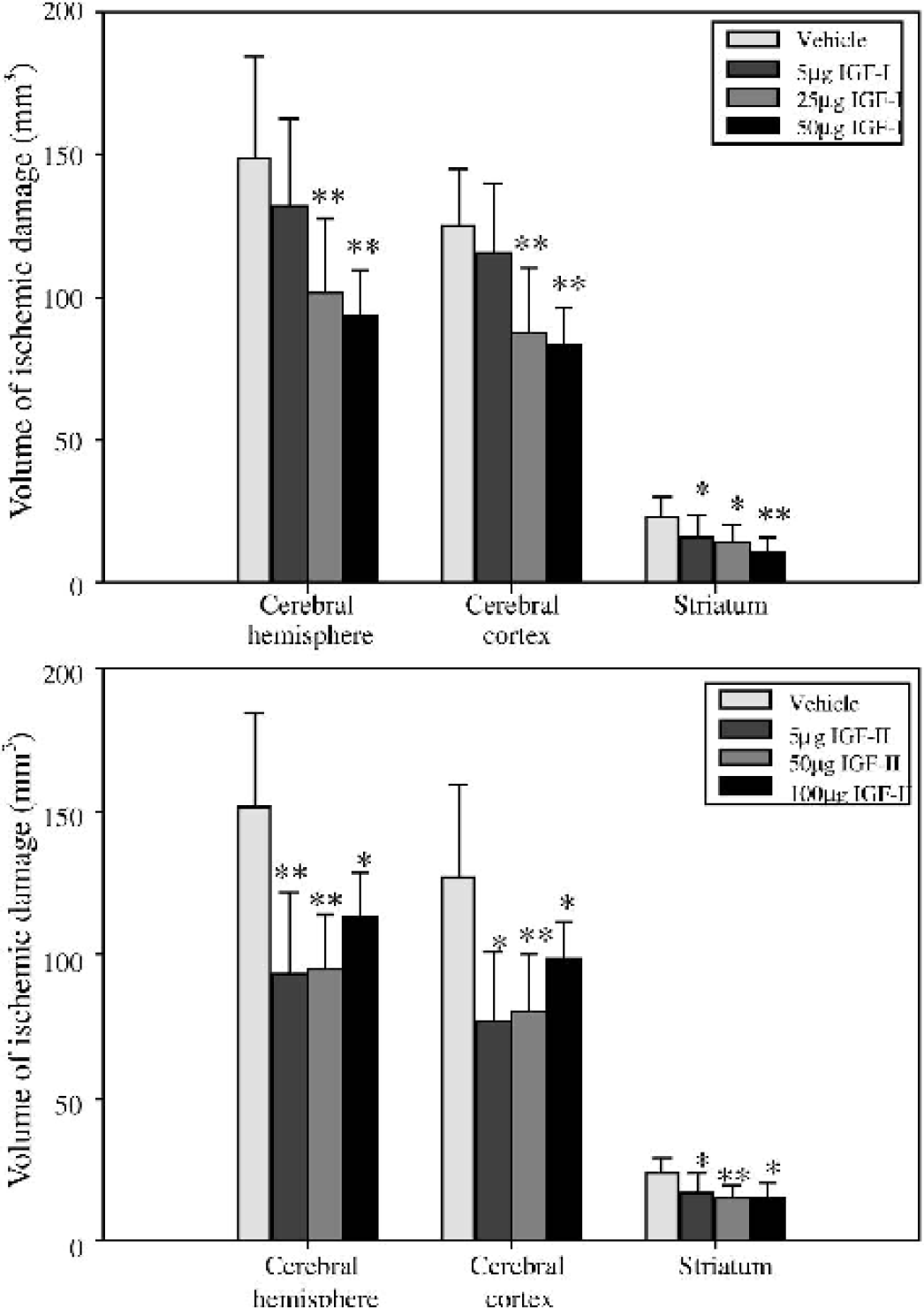
Effect of IGF-I
All doses of IGF-II tested resulted in a significant reduction of the total, cortical, and striatal lesion volume (Fig. 3). The highest dose (100 μg) appeared to cause the smallest reduction of the total lesion volume (25%), compared with the 5 μg (38%) and 50 μg (37%) doses.
All doses of NBI-31772 tested in the subtemporal model of stroke resulted in a significant reduction of the total and cortical lesion volume (Fig. 4). The two higher doses (50 and 100 μg) had a similar effect on the total lesion volume that was slightly greater than that of the 5 μg dose (40% reduction, compared with 31%). Only the 50 μg dose had a significant effect on the striatal lesion volume (40% reduction, P < 0.05). These results were similar to those observed in a previous study (60 μg NBI-31772 caused a 44% reduction of the total lesion volume, data not shown).
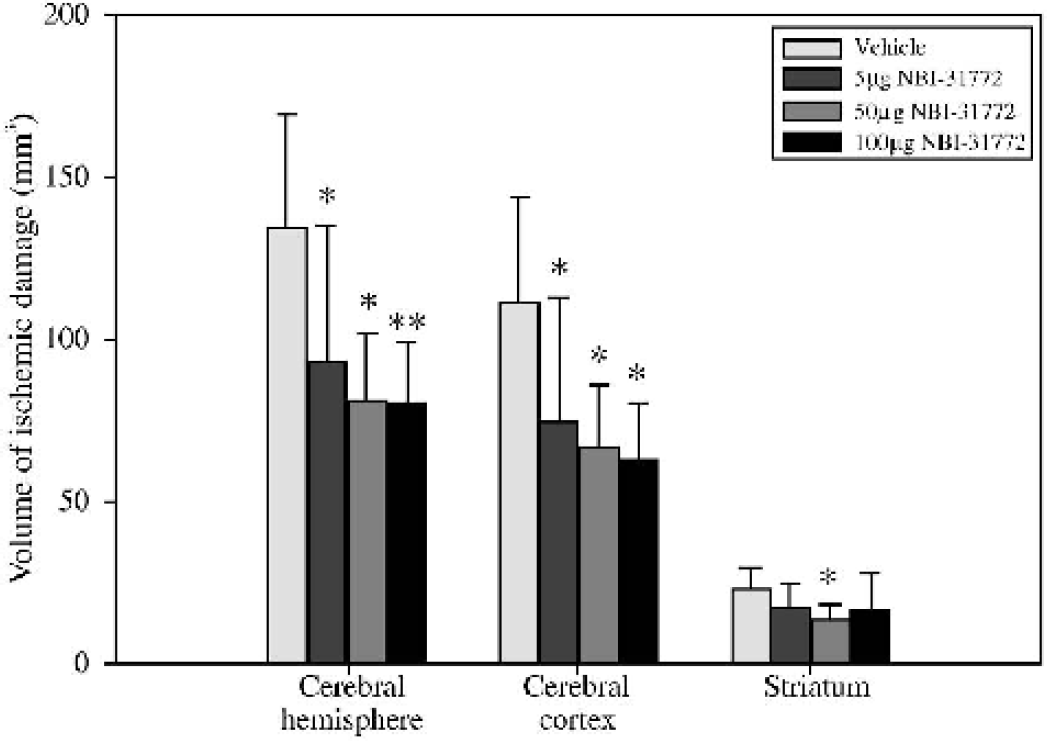
Effect of NBI-31772 on the volume of ischemic damage assessed 24 hours after permanent transcranial MCAO in the rat. Animals were injected icv with NBI-31772 (5, 50, or 100 μg) or vehicle (water, 5 μL) immediately after MCAO. Data are mean ± SD (n = 5 to 9 per group). ∗ P < 0.05, ∗∗ P < 0.01, compared with vehicle-treated group (analysis of variance followed by Dunnett's test). MCAO, middle cerebral artery occlusion; icv, intracerebroventricular.
Intraluminal occlusion of the MCA via a cervical carotid approach produced an ischemic lesion that was larger than that observed following subtemporal MCAO at 24 hours. Ischemic damage was observed not only within the territory of the occluded MCA (dorsolateral cortex and striatum) but also in brain areas outside the vascular territory of the MCA, that is, the hypothalamus, thalamus, septum, and midline cortex. NBI-31772 (50 μg, injected icv at the time of MCAO) had a similar neuroprotective effect in the intraluminal model of stroke (40% reduction of the cortical lesion volume, P < 0.001), although there was no reduction of the striatal lesion (Fig. 5). NBI-31772 was still effective when administered 3 hours after the induction of ischemia (18% reduction of total lesion volume, P < 0.05). The effect of NBI-31772 on ischemic volume was dependent upon the time of administration, with the greatest reduction in the lesion being observed when drug was given immediately after MCAO (Fig. 5). The effect of NBI-31772 on brain swelling was also dependent upon the time of administration (Fig. 5), and a significant effect was observed only when drug was administered immediately after MCAO (24% reduction, P < 0.05).
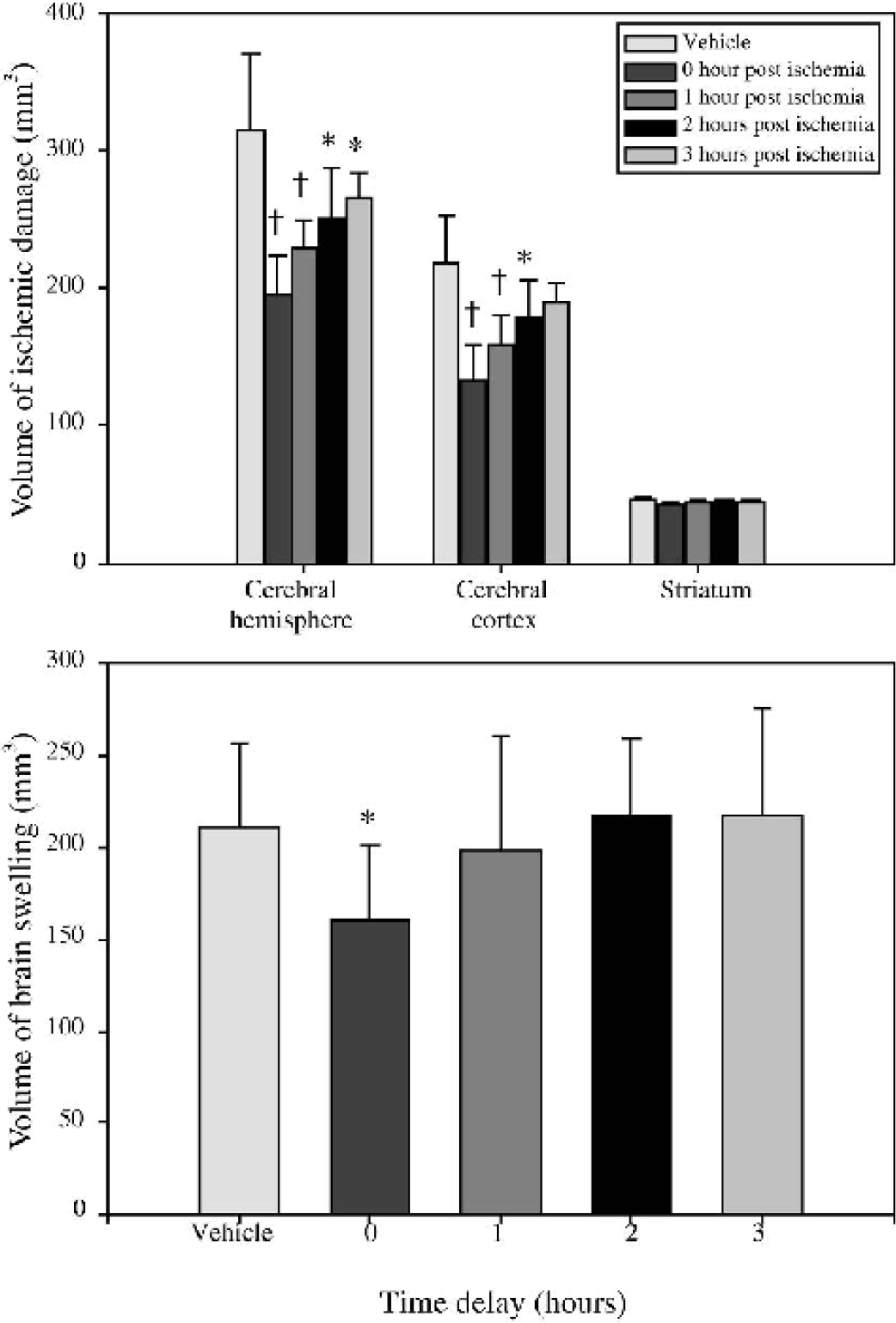
Therapeutic time window for NBI-31772 against volume of ischemic damage
There was a good linear relationship between the volume of ischemic damage and the volume of brain swelling which was similar for vehicle-treated animals and all the NBI-31772 treated groups irrespective of the time delay of treatment onset, with an overall correlation coefficient r = 0.612 (P < 0.001).
DISCUSSION
The results of the present study demonstrate the neuroprotective properties of the IGF system in experimental models of ischemia in vitro and in vivo. Treatment with IGF-I, IGF-II, and IGFBP-LI protected CA1 neurons against ischemic-like injury in hippocampal slice cultures. Exogenously administered IGF-I and IGF-II significantly reduced the volume of ischemic damage after permanent focal cerebral ischemia in the rat. Administration of NBI-31772, a small molecule which binds to endogenous IGFBPs and releases biologically active IGF (Chen et al., 2001; Liu et al., 2001b), elicited a neuroprotective effect similar to that of IGF itself and was effective when administered up to 3 hours after permanent occlusion of the MCA. These results extend and substantiate the authors' previous study (Loddick et al., 1998) and suggest that the pharmacologic displacement of IGF from its binding proteins may be a useful therapeutic strategy for the treatment of stroke.
Neuroprotective effects of IGF-I have been described previously in a variety of in vitro and in vivo forms of neuronal injury. Studies in vitro have demonstrated that IGF-I can protect a variety of neuronal cell types (i.e., cortical, hippocampal, septal, cerebellar granule, and hypothalamic) from various different insults (i.e., glucose deprivation, calcium-induced hypoglycemia, oxidative stress, serum withdrawal, and β-amyloid–induced injury) (Cheng and Mattson, 1992; Galli et al., 1995; Sortino and Canonico, 1996; Dore et al., 1997; Tagami et al., 1997; Venters et al., 1999; Nicholas et al., 2002). In vivo, protective effects of IGF-I have been described in models of global and hypoxic ischemia (Cheng and Mattson, 1992; Zhu and Auer, 1994; Guan et al., 1996; Johnston et al., 1996; Guan et al., 2000b; Guan et al., 2001) and more recently, in models of stroke (temporary and permanent focal cerebral ischemia) (Loddick et al., 1998; Wang et al., 2000; Schabitz et al., 2001; Liu et al., 2001a). Data from the present study confirm and extend these previous observations by demonstrating the dose-dependent nature of the neuroprotective effects of IGF-I, and in addition, they demonstrate for the first time similar neuroprotective effects of IGF-II in permanent focal cerebral ischemia.
In comparison with the numerous studies on IGF-I, the effect of IGF-II on neuronal death is less described, with only a few studies reported in the literature. Protective effects of IGF-II have been described in vitro on cultured oligodendrocytes and hippocampal and septal neurons (Cheng and Mattson, 1992; Nicholas et al., 2002). However, the only published in vivo study found no evidence of a protective action of IGF-II in the brain. In fact, this study reported that whereas IGF-I reduced brain damage after hypoxic ischemic injury, coadministration of IGF-II with IGF-I attenuated this neuroprotective effect (Guan et al., 1996). The authors proposed that this effect of IGF-II was caused by competition for a transport/translocation mechanism that was necessary for the neuroprotective effects of IGF-I. The authors suggested that IGFBPs might act as the transport mechanism and subsequently demonstrated that IGF-I and IGFBP-2 are colocalized in the brain after hypoxic ischemic injury (Guan et al., 2000a). Our present results are not consistent with this hypothesis. In contrast to the previous study in hypoxic ischemia, IGF-II reduced the volume of brain injury after permanent focal cerebral ischemia. With icv administration, IGF-II was as effective as IGF-I at reducing the amount of brain injury (37% reduction of the total lesion volume) but at a lower dose (5 μg) than IGF-I (50 μg). This result is somewhat surprising given that IGF-II has a lower affinity for the IGF-1 receptor than IGF-I, although IGF-I and IGF-II have an almost identical dose-response curve in an in vitro study of hypoglycemia-induced neuronal death (Cheng and Mattson, 1992). The present results may suggest that the neuroprotective effects of IGF-I and IGF-II are not mediated by the known IGF-1 receptor. However, it is also possible that the apparent difference in potency is caused by differences in the rate of translocation/transport of the IGFs into brain parenchyma to their site of action.
The effects of the IGF peptides were evaluated in a model of OGD-induced cell death in organotypic hippocampal slice cultures. The RT-PCR analysis demonstrating that the mRNA of the expected members of the IGF family were expressed in the hippocampus of the organotypic cultures would suggest that this is a suitable model system in which to explore the neuroprotective effects of IGF peptides in vitro. Administration of the ischemic hippocampal slice cultures with IGF peptides prevented neuronal cell death, with the rank order of neuroprotection in this isolated cell-based brain ischemia model being IGF-I > IGF-II > IGFBP-LI. The expression profile of the RT-PCR data indicate that IGF-I, IGF-II, and many of the IGF-BPs are endogenously expressed by the organotypic hippocampal slices on day 14 of culture, suggesting that the neuroprotective effect of IGFBP-LI may be a result of dissociating IGF peptides from the binding protein present in both the slice cultures and culture media.
The mechanism of transport of IGFs is unclear. IGFs can enter the brain from the periphery, by a mechanism that appears to be carrier mediated. However the transport of IGF across the blood brain barrier has been shown to be independent of either IGFBPs or IGF receptors (Armstrong et al., 2000; Pulford and Ishii, 2001), whereas it has been suggested that the transport of IGFs from CSF into brain parenchyma of hypoxic ischemic brains is dependent on IGFBPs (Guan et al., 1996; Guan et al., 2000a). The present results along with the authors' previous study (Loddick et al., 1998) suggest that IGFBPs are not essential for the neuroprotective effects of IGF in permanent focal cerebral ischemia. In the present study, icv administration of NBI-31772, a nonpeptide, small molecule that binds to the IGFBPs and displaces IGFs, was as effective as IGF-I and IGF-II at reducing the volume of brain injury after permanent focal cerebral ischemia. As this compound has no activity at the IGF receptors, this neuroprotective effect is presumably mediated by the increase in free biologically active IGF within the brain. NBI-31772 was equally effective in the two models of experimental stroke despite the difference in the magnitude of the total lesion volume in the two models. NBI-31772 was effective at reducing the lesion volume even when administration was delayed until 3 hours after MCAO, suggesting a relatively long therapeutic window. These results are consistent with a previous study that used magnetic resonance imaging to demonstrate a delayed neuroprotective effect of IGF-I in transient focal cerebral ischemia, with no difference in cerebral blood flow, compared with the vehicle-treated control group (Schabitz et al., 2001). These authors also reported that the neuroprotective effects of IGF-I were sustained for 7 days after ischemia, suggesting a permanent neuroprotective effect.
CONCLUSIONS
The present data demonstrate that elevated levels of IGFs are neuroprotective in in vitro and in vivo models of cerebral ischemia. Moreover, the finding that pharmacologic displacement and elevation of endogenous, bioactive IGFs from IGFBPs by peptide and small molecule IGFBP ligand inhibitors can confer benefit when given after the onset of permanent MCAO in two models of focal ischemia (Loddick et al., 1998; present study) is of particular note. Displacement of endogenous IGFs from IGFBPs in the ischemic brain may offer a therapeutic advantage over treatment with IGF itself. The beneficial effects of IGF-I directly may be limited by the presence of elevated levels of IGFBPs in the injured brain. Further, this strategy may produce fewer side effects than treatment with IGF-I itself, since the maximum amount of free IGF released to interact with IGF receptors will be limited by the total amount of endogenous IGF in the brain. Finally, the observation that the small molecule IGFBP ligand inhibitor NBI-31772 is beneficial when given icv suggests that it may be possible to develop future small molecule IGFBP ligand inhibitors that are neuroprotective when given systemically and which may be used for the treatment of stroke and other neurodegenerative disorders.
Footnotes
Acknowledgments
The authors thank Anthea Hughes and Dacie Lewis for performing the icv cannulations, Joelle Eggold for RT-PCR analysis, and Dr. Nick Ling and Dr. Chen Chen from the Departments of Peptide and Medicinal Chemistry for the synthesis of IGFBP-LI and NBI-31772.