
Abstract
The no-reflow phenomenon and delayed hypoperfusion after transient cardiac arrest (CA) impede postischemic recovery. Activation of lipid peroxidation (LPO) after ischemia and reperfusion is considered one of the mechanisms responsible for such abnormalities. The present study investigates the influence of iron-dependent LPO inhibitor deferoxamine (DFO) on the cerebral perfusion after prolonged CA and resuscitation. Fourteen male Sprague-Dawley rats were subjected to 17 minutes of CA, induced by esmolol (an ultrashort-acting β-blocker) and apnea, followed by resuscitation by retrograde intraaortic infusion of oxygenated donor blood mixed with a resuscitation cocktail inside a vertical-bore 9.4-T magnetic resonance imaging (MRI) magnet. Animals were randomized double-blindly into two groups to receive DFO or saline, respectively. Cerebral perfusion was measured by MRI continuously using the arterial spin-labeling method before, during, and after CA. All animals were successfully resuscitated in 1.36 ± 0.04 minutes with well-controlled arrest time (17.99 ± 0.03 minutes) in both groups. Deferoxamine significantly increased cerebral perfusion in hippocampus, thalamus, hypothalamus, and amygdala, but not in cortex, during the first 20 minutes of reperfusion. In the DFO-treated group, the neurologic deficit score was significantly better (400 ± 30 vs. 250 ± 47, out of 500 as the best, P < 0.05) and weight loss was significantly less (33 ± 6 vs. 71 ± 19 g, P < 0.05) 5 d after arrest. The finding supports the notion that early reperfusion immediately after resuscitation is important for long-term outcome and that LPO may be involved in microvascular disorders during the reperfusion, particularly in the brain after prolonged cardiac arrest and resuscitation.
Keywords
Cardiac arrest (CA) leads to global brain ischemia. The development of abnormalities in cerebral perfusion, notably the no-reflow phenomenon and hypoperfusion at the early stage of reperfusion after resuscitation, is one of the determinative factors for poor outcome in postischemic recovery (Böttiger et al., 1997; Fischer and Hossmann, 1995; Liachenko et al., 2001). Activation of the free radical reactions in the form of lipid peroxidation (LPO) after ischemia and reperfusion is considered one of the mechanisms responsible for such abnormalities (Chan, 2001; Moore and Traystman, 1994). Initiation of LPO cascade involves soluble iron ions through the decomposition of preformed lipid peroxides, and possibly the generation of the most detrimental hydroxyl radical through the Fenton and Haber-Weiss chemistry (Babbs, 1985; Schaich and Borg, 1988). Active soluble iron ions, required for LPO initiation, are predominantly supplied by a cytosolic pool of labile low-molecular-weight iron (LMWI) (Jacobs, 1976), which increases dramatically under the ischemic conditions (Voogd et al., 1992). Decrease in intracellular pH (Hurn et al., 1995) in the presence of superoxide radicals causes fast mobilization of iron from its storage (ferritin) and transport (transferrin) systems to LMWI pool (Biemond et al., 1988).
Low-molecular-weight iron is accessible for scavenging by externally administered iron chelators, such as ethylenediaminetetraacetic acid, deferoxamine (DFO), and dextrazoxane. These chelators can, therefore, be used to attenuate iron toxicity (Elihu et al., 1998). Beneficial effects of iron chelation therapy have been shown in different in vitro and in vivo ischemia models of brain (Hurn et al., 1995; Palmer et al., 1994), kidneys (Gower et al., 1989), liver (Drugas et al., 1991), and other organs and tissues (Shibuya et al., 1996). For example, DFO has been applied to the treatment of reperfusion consequences after global and partial ischemia and used as a protective agent in the storage and preservation of organs. Addition of DFO to rinsing solution reduced peroxidative damage of isolated lungs (Shibuya et al., 1996). Cardioplegic solution with DFO showed additional protection against reperfusion injury to the heart (Nicholson et al., 1997). Systemic administration of DFO before removal of kidney for transplantation adds additional protection against damage caused by LPO during storage (Gower et al., 1989). In vivo administration of DFO has been shown to improve muscle contractile function after reperfusion after tourniquet ischemia (Zavitsanos et al., 1996), reduce pathologically enhanced leukocyte adhesion, and improve hepatic microcirculation after resuscitation from hemorrhagic shock (Bauer et al., 1999), and attenuate liver injury in endotoxinenhanced liver injury after hepatic ischemia (Drugas et al., 1991). In various models of cerebral ischemia, DFO has also been shown to decrease the amount of damage caused by free radicals. Low-molecular-weight DFO can help the recovery of cerebral pH, ATP, and phosphocreatine after incomplete brain ischemia caused by increased intracranial pressure with elevated plasma glucose concentration (Hurn et al., 1995), and decrease morphologic damage after cerebral hypoxia–ischemia in neonatal rats (Palmer et al., 1994). Hydroxyethyl starch–conjugated DFO has been shown to reduce neurologic injury and the accumulation of LPO products (Rosenthal et al., 1992). In many cases, beneficial effect of DFO can be attributed principally to its iron chelation properties, while direct radical scavenging may also be involved (Halliwell, 1989; Hurn et al., 1995; Palmer et al., 1994; Sarco et al., 2000).
The present study investigates the influence of DFO on the cerebral reperfusion and outcome after resuscitation after prolonged cardiac arrest. A newly developed model of cardiac arrest and resuscitation was used (Liachenko et al., 1998, 2001; Xu et al., 2002) in conjunction with noninvasive magnetic resonance imaging measurements of the cerebral perfusion.
MATERIALS AND METHODS
The experimental protocol was approved by our Institutional Animal Care and Use Committee. Fourteen male Sprague-Dawley rats (220 ± 9 g; Harlan Sprague-Dawley, Inc., Indianapolis, IN, U.S.A.) were used. Animals were subjected to 17 minutes of cardiac arrest caused by intravenous esmolol injection and apnea under normothermic conditions. Resuscitation was achieved by retrograde intraaortic infusion of resuscitation mixture (Liachenko et al., 1998, 2001). Rats were randomized double-blindly into two experimental groups, treated with saline (control) and DFO, respectively. Cerebral perfusion was measured by continuous magnetic resonance imaging perfusion mapping using the arterial spin labeling method. Survival, histologic deficit score (HDS), neurologic deficit score [NDS, as detailed in (Liachenko et al., 2001; Neumar et al., 1995)], and body weight changes were evaluated 5 d after resuscitation.
Animal preparation
Animals were prepared as described previously (Liachenko et al., 1998, 2001). Surgery was performed with the animals under general anesthesia (isoflurane, 1.25%) and mechanical ventilation (50:50, N2O to O2, 1 mL per 100 g body weight, 40 strokes per minute, and 5-cm H2O positive end-expiratory pressure) after orotracheal intubation and muscle relaxation (pancuronium bromide, 2 mg/kg). One femoral vein and two femoral arteries were catheterized with biocompatible Micro-Renathane (venous) and RenaPulse (arterial, Braintree Scientific, Inc., Braintree, MA, U.S.A.) tubing. Arterial blood pressure was monitored continuously throughout the experiment. Rats were snugly placed in a custom designed 8-element birdcage probe (inner diameter = 44 mm) (Xu and Tang, 1997) and positioned inside the 9.4-T vertical-bore magnet of a Varian/Chemagnetics CMXW-400SLI imaging spectrometer. Once the animals were inside the magnet, the anesthesia and ventilation parameters were adjusted as detailed previously (Liachenko et al., 1998, 2001; Xu et al., 2002). The rat body temperature, measured by a rectal temperature probe (YSI 402; Yellow Springs Instruments, Yellow Springs, OH, U.S.A.), was maintained at 36.6 ± 0.1°C with a feedback-controlled air-heating blanket.
Cardiac arrest and resuscitation
Cardiac arrest started with injection of esmolol (6.25 mg, intravenously) 5 minutes after short-action paralysis with 1 mg/kg vecuronium bromide (Liachenko et al., 2001). Complete cessation of blood flow (defined as a drop of mean arterial blood pressure (MABP) to <12 mm Hg, and pulse pressure to <5 mm Hg) was observed in 22 ± 1 seconds. Mechanical ventilation, isoflurane anesthesia, and heating of the animal were discontinued during the time of CA. Resuscitation begun after 17 minutes of CA with restoration of the ventilation and the infusion into the femoral artery of donor arterial blood mixed with epinephrine (8 μg/mL), sodium bicarbonate (0.05 mEq/mL), and heparin (5 U/mL) (Liachenko et al., 1998, 2001). Infusion was performed manually to maintain the systemic MABP at 26 ± 5 mm Hg. At the first spontaneous cardiac contraction (monitored on real-time blood pressure display), the infusion of resuscitation mixture was stopped, and return of spontaneous circulation (ROSC) followed shortly thereafter. The rats were then continually ventilated for 3 or more hours with anesthesia reinstated as required. At least 3 h after resuscitation, rats were taken out of the magnet and detached from all catheters and ventilator as described previously (Liachenko et al., 1998, 2001).
Deferoxamine administration
Rats were randomized into two groups double-blindly to receive either DFO or vehicle as preresuscitation treatment. The control group (n = 7) received a bolus of physiologic saline (2 mL/kg, intravenously) 5 minutes before resuscitation. The drug-treated group (n = 7) received a dose of 100 mg/kg of DFO in equivalent amount of saline also 5 minutes before resuscitation. Deferoxamine solution was prepared by dissolving deferoxamine mesylate (Sigma Chemical Co., St. Louis, MO, U.S.A.) in sterile saline to a concentration 50 mg/mL. The double-blind design was achieved as follows. Investigator 1 freshly prepared both solutions on the dates of experiments, and labeled them as solution A and B. Investigator 2, who had no knowledge of the identity of A and B, randomly selected A or B solution for treatment without revealing the solution labels to other investigators. The minimum number of experiments for each group, calculated for a power of 0.8 with significance α of 0.05 and anticipated difference of at least twice the standard deviation of the measurements, is 6. Randomization was continued until n ≥ 6 and A and B solutions were used for the same number of times. The keys to A and B and the solution identity used for each experiment were not revealed until all experiments were done and all data were completely processed.
Outcome evaluation.
Postoperative animal care and outcome evaluation were performed as described previously (Liachenko et al., 1998, 2001; Xu et al., 2002). Animals were kept under the standard environmental conditions in individual cages and under constant observation. No measurements of body temperature or other physiologic variables were performed during this period. After 5 d of observation, the final NDS was evaluated using the criteria described previously (Liachenko et al., 2001; Neumar et al., 1995). The scoring system ranges from 0 for brain dead to 500 for neurologically normal. Animals were then anesthetized with isoflurane and their brains were perfused with buffered 10% formalin and processed further for conventional hematoxylin–eosin staining (Liachenko et al., 1998). Histologic damage was evaluated by counting the total and ischemic neurons (Liachenko et al., 1998, 2001) in the CA1 region of the hippocampus on digital microphotographs using Adobe Photoshop software (Adobe Systems, Inc., San Jose, CA, U.S.A.). Histologic deficit score was expressed as a percentage of ischemically changed neurons against the total. The NDS and HDS values were assessed by the same investigator, blinded to the particulars of the experiment.
Magnetic resonance imaging perfusion measurement
Magnetic resonance imaging experiments were performed to evaluate the cerebral perfusion during and after cardiac arrest and resuscitation using the arterial spin labeling method as described previously (Liachenko et al., 2001; Xu et al., 2002). Imaging parameters were as follows: image plane at the level of dorsal hippocampus [brain level 30 to 32 (Swanson, 1998)], tagging plane offset = ± 14 mm, tagging power ≈500 mW, which was adjusted to provide the degree of spin inversion (the α value) at 0.48 ± 0.01 before CA (Liachenko et al., 2001), tagging duration = 0.8 seconds, spin-echo time = 14 milliseconds, repetition time = 1 second, number of average = 2, slice thickness = 3 mm, matrix size = 128 × 64 and zero-filled to 128 × 128 before Fourier transform, and field of view = 44 × 44 mm2. Perfusion maps were calculated pixel-by-pixel as described earlier (Liachenko et al., 2001; Xu et al., 2002). Perfusion images were acquired continuously during CA and after resuscitation, with a temporal resolution of 2.5 minutes per perfusion map.
Data analysis
Perfusion maps were processed with individually adjusted anatomic masks using the customized LabVIEW software (National Instruments, Inc., Austin, TX, U.S.A.) as detailed before (Liachenko et al., 1998, 2001; Wang et al., 2002; Xu et al., 2002; Yushmanov et al., 2002) and expressed as relative changes (in percent) over time against the averaged preischemic values in the corresponding brain regions. Statistical analysis was performed using the SPSS program (SPSS, Inc., Chicago, IL, U.S.A.). Independent samples t-test was used to compare variables of control and DFO-treated groups. Non-parametric Mann-Whitney test was used to compare NDS and HDS values. Cerebral perfusion 5 d after resuscitation in the surviving animals was compared with the preischemic level using the paired t-test. Repeated-measures design of general linear model of analysis of variance (ANOVA) was used to determine the effect of DFO on perfusion and cardiovascular performance over time. Survival of animals was analyzed using Fisher's exact test and Kaplan-Meier method with censored values set at 120 h for surviving rats. All data are reported as mean ± SEM unless stated otherwise.
RESULTS
Cardiac arrest and resuscitation were highly reproducible in both groups. Using the Utstein-style guidelines for uniform reporting laboratory cardiopulmonary resuscitation research (Idris et al., 1996), the arrest time (nonintervention interval) in the present study was 17 minutes, and the duration between retrograde intraaortic infusion and ROSC was 1.36 ± 0.04 minutes. All animals were successfully resuscitated. The duration of circulatory arrest (interval between the cessation of blood flow and ROSC) among all animals was 17.99 ± 0.03 minutes. The average amount of epinephrine received by animals during resuscitation was 74.0 ± 2.5 μg/kg with no difference between groups. Parameters of CA and resuscitation in control and DFO-treated groups are listed in Table 1. There were no significant differences in the arrest and resuscitation times between two groups. At the end of the observation period, five animals (71%) were alive in the DFO-treated group and only 3 (43%) survived in the control group. However, the mean survival time, censored at 120 h and without considering the neurologic deficits of the animals at 120 h, is not significantly different between the groups. Surviving animals in the DFO-treated group showed significantly better NDS and less weight change than in the control group. There was no difference in histologic damage between groups. The average percentage of ischemically changed neurons in the CA1 region of the hippocampus of all surviving rats was 66 ± 8%.
Parameters of cardiac arrest and long-term outcome

Data are presented as median (minimum, maximum) for NDS and HDS and as mean ± SEM elsewhere. n = number of animals studied (number of animals that survived for 5 d). Mean survival time was calculated using Kaplan-Meier survival test with censored values (survived animals) restricted to 120 h.
Significant difference between groups (P < 0.05, Mann-Whitney test)
significant difference between groups (P < 0.05, t-test).
NDS, neurologic deficit score; HDS, histologic deficit score; DFO, deferoxamine.
The recordings of arterial blood pressure in all animals are nearly identical to that in Fig. 2 of our previous publication (Liachenko et al., 2001), except that the duration of apnea is now 17 minutes. Within 22 seconds after esmolol injection, the MABP dropped to less than 12 mm Hg and the arterial pulse pressure was less than 5 mm Hg, at which point the onset of circulatory arrest was defined. The MABP during the arrest time ranged from 12 to 0 mm Hg with a median of less than 1 mm Hg. The pulse pressure during this period was too small to accurately measure the heart rate. Changes in MABP and heart rate in response to approximately 17 minutes of CA and subsequent resuscitation, averaged among animals within the same group, are plotted in Figs. 1A and 1B, respectively. Both MABP and heart rate (HR) recovered more rapidly in the control group during first 5 minutes after ROSC (F = 4.949, P = 0.046, for MABP and F = 88.391, P < 0.001, for HR by repeated-measures ANOVA). At the later stage of reperfusion, there was no significant difference between groups (F = 1.229, P = 0.289, for MABP and F = 3.304, P = 0.094, for HR by repeated-measures ANOVA during the period of 5 to 20 minutes after ROSC).
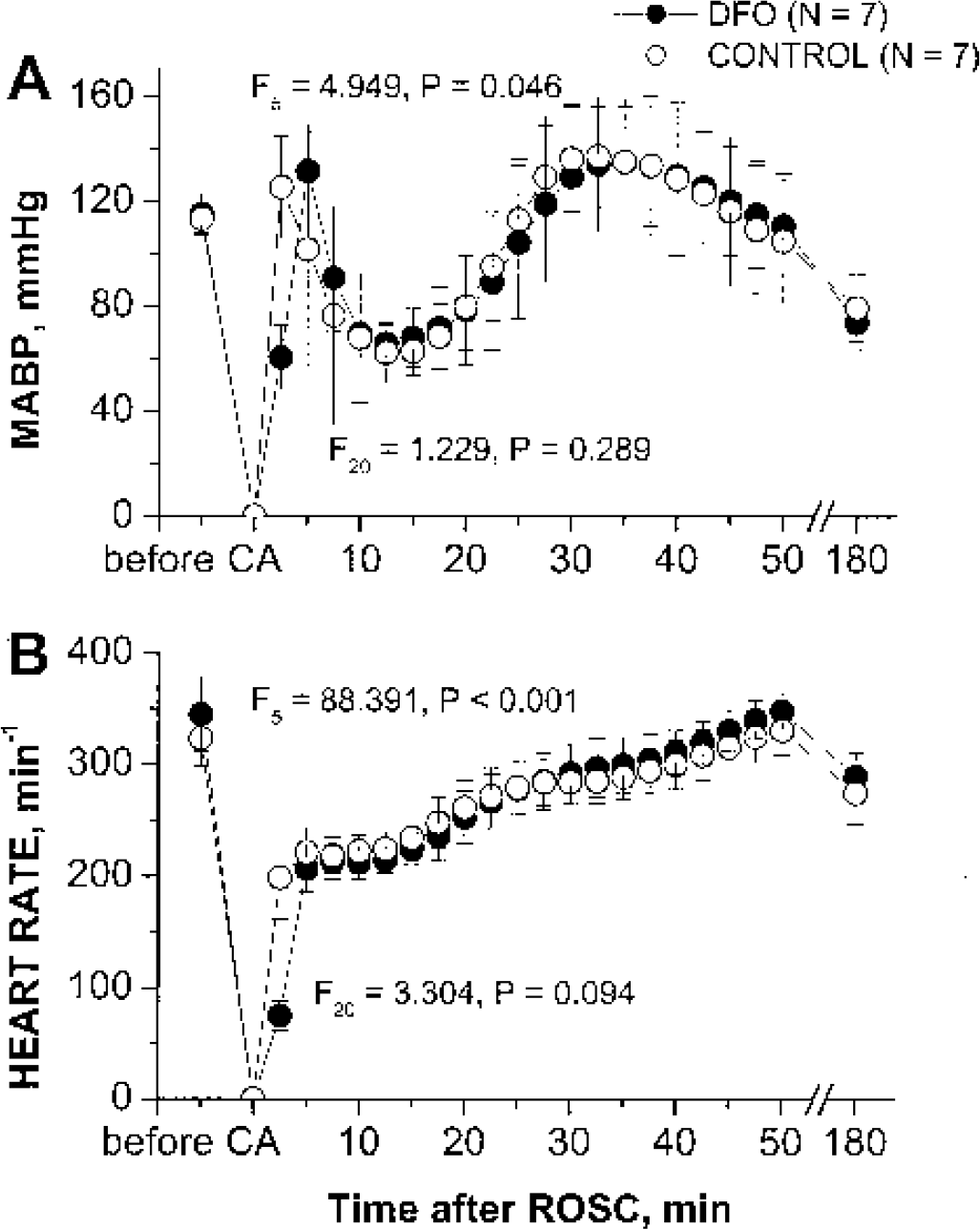
Effect of deferoxamine (DFO) on the time course of mean arterial blood pressure (MABP)
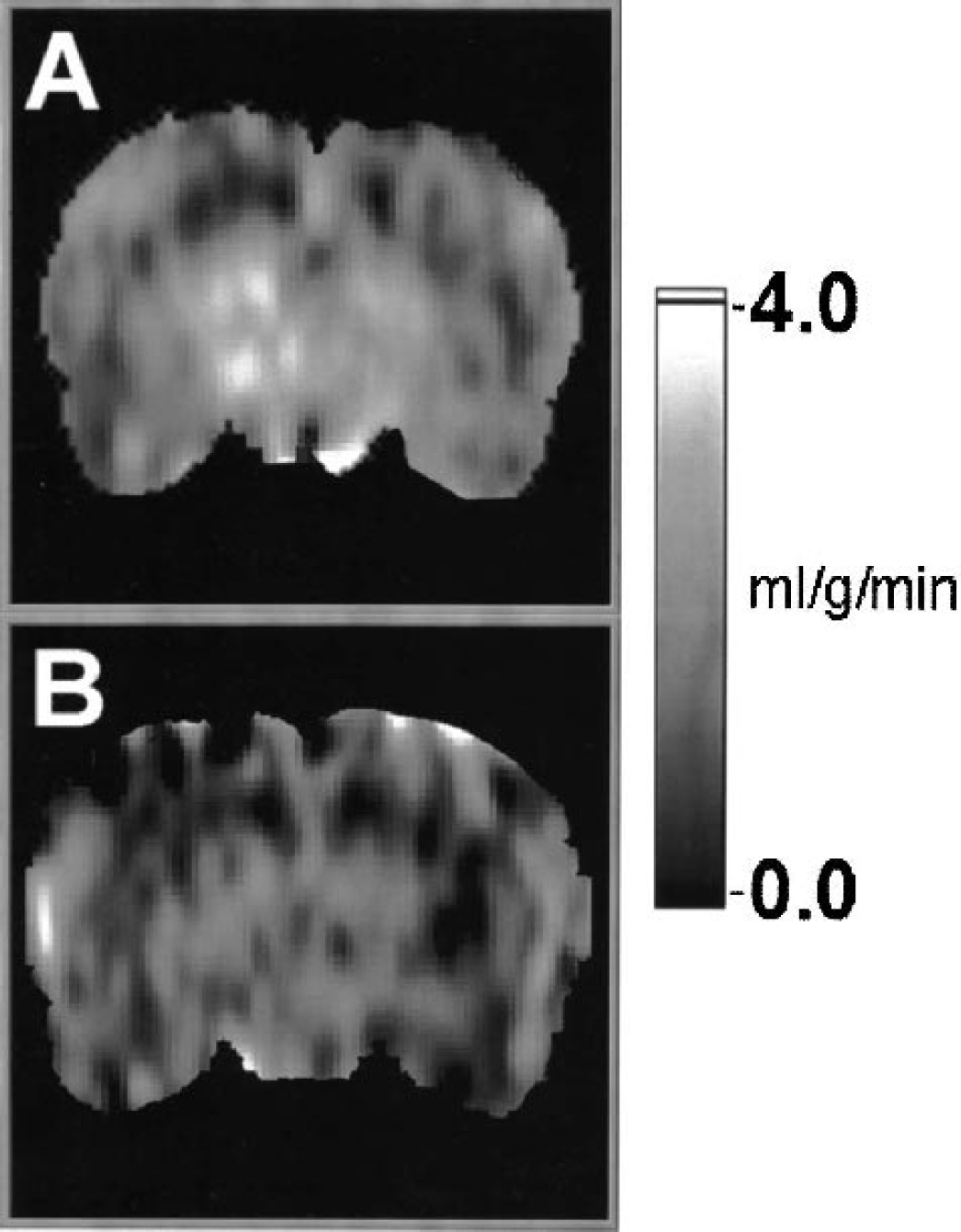
Representative magnetic resonance imaging cerebral perfusion maps of rats in the deferoxamine-treated
Figure 2 depicts representative cerebral perfusion maps of rats in the DFO-treated (Fig. 2A) and control (Fig. 2B) groups 10 minutes after ROSC. The changes in animal-averaged perfusion over time after CA in different brain structures are plotted in Fig. 3. Animals treated with DFO showed significantly higher cerebral perfusion during the period of 5 to 20 minutes after resuscitation in hippocampus, thalamus, hypothalamus, and amygdala, but not in cortex. Overall brain-averaged perfusion during this period was also higher in the DFO-treated group. There was no significant difference in cerebral perfusion between groups either during the first 5 minutes or after 20 minutes of reperfusion. Fifty minutes after resuscitation, all animals developed protracted hypoperfusion (15% to 30% of preischemic cerebral perfusion level). Averaged cerebral blood flow restored to 50% to 75% of preischemia level at 3 h after resuscitation. Five days after resuscitation, cerebral perfusion in surviving animals did not differ significantly from normal values, and no differences between groups were detected (t = 0.132, P = 0.900 by two-sample t-test).
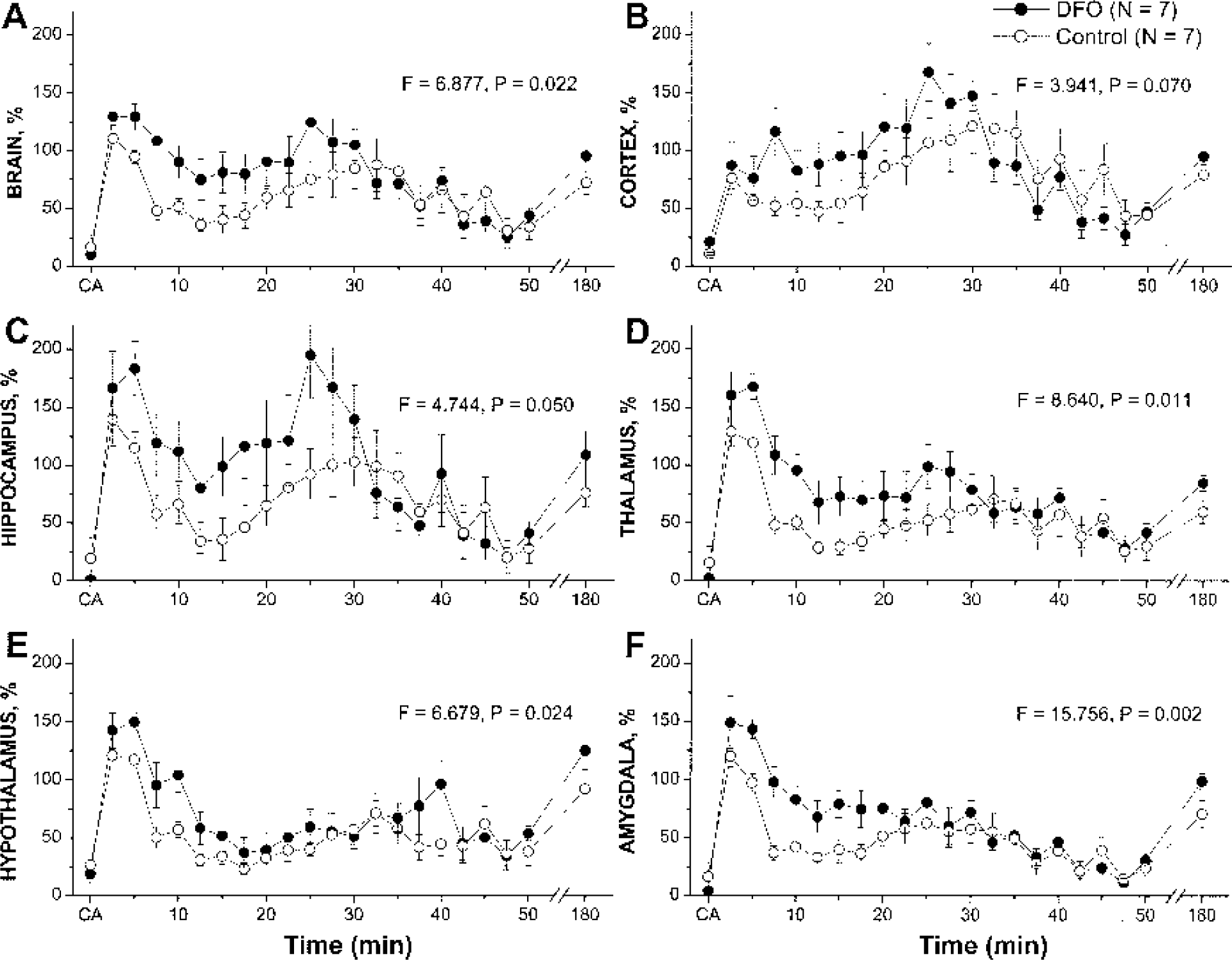
Effect of deferoxamine (DFO) on the time course of reperfusion in different brain structures of rats after resuscitation from 17 minutes of cardiac arrest.
DISCUSSION
In the present study, we showed the effect of preresuscitation administration of DFO on the enhancement of postarrest cerebral perfusion, which was accompanied by somewhat better neurologic scores. This finding further supports the hypothesis that inhibition of iron-dependent LPO plays certain roles in the amelioration of microvascular disorders after global cerebral ischemia (Hein and Kuo, 1998; Luo et al., 1995). Recently, we demonstrated (Xu et al., 2002) that resuscitation efficacy had profound effects on the initial reperfusion, which in turn had determinative effects on the long-term outcome. High perfusion during approximately the first 20 minutes of reperfusion by effective resuscitation led to much better outcome than did the poor resuscitation and low initial reperfusion. We also showed that the dependence of outcome on the initial cerebral reperfusion is relatively insensitive to the duration of CA. A long period of CA does not necessarily lead to worse long-term outcome than a short period of CA as long as initial reperfusion is restored adequately. In the present study, we used a pharmacologic means to enhance the initial reperfusion after a very long period of no-flow. We reached the same conclusion, that initial reperfusion is of determinant importance for long-term survival after cardiac arrest and resuscitation. However, it should be pointed out that other protective mechanisms of DFO injection, such as osmotic load or possible direct vasodilation, might also contribute to the observed effects. Moreover, the current experimental design does not allow us to determine the direct correlation between DFO-induced alleviation of hypoperfusion and the probability of long-term animal survival.
Cardiac arrest and resuscitation, similar to other causes of ischemia and reperfusion, are known to instigate the so-called “oxidative burst,” which is characterized by the surge of free radical reaction cascade, particularly LPO (Babbs, 1985; Chan, 2001). The initiation of LPO cascade involves the generation of mild and highly reactive oxygen radical species, including superoxide anion radical, hydroperoxide, and hydroxyl radical. The generation of the most deleterious hydroxyl radical (•OH) requires the participation of transition metal ion that can catalyze an electron transfer in Fenton and Haber-Weiss reactions (Babbs, 1985; Schaich and Borg, 1988). The best candidate for the role of catalytic transition metal is cytosolic iron, not bound to large organic complexes such as porphyrins (e.g., hemoglobin). There is a sufficiently high amount of nonheme iron in the brain tissue (Hill and Switzer, 1984; Sarco et al., 2000), part of which exists in a catalytically active reduced ferrous state (Fe2+) as complexes with low-molecular-weight ligands. The majority of nonheme iron in the brain, as in other tissues, is trapped in specialized proteins: ferritin for storage or transferrin for transport. After ischemia and reperfusion insults, a large amount of iron is released into a transient pool of LMWI (Krause et al., 1985; Lipscomb et al., 1998). Superoxide and decreased cytosolic pH are believed to participate in postischemic iron release from ferritin (Lipscomb et al., 1998). It has been shown that postischemic reperfusion is the necessary step to induce intracellular LMWI accumulation (Krause et al., 1985).
The iron in the transient LMWI pool is readily available for initiation and propagation of LPO processes. This pool can be trapped by externally administered metal chelators, justifying the iron chelation therapy of ischemia and reperfusion injuries as well as other pathologies with enhanced level of LPO (Halliwell, 1989). Deferoxamine is a well-known substance with the ability to bind iron ions to form neutral iron complexes, which cannot catalyze LPO reactions. In addition to iron chelator properties, DFO has the ability to directly scavenge free radicals when applied in relatively high concentrations (Halliwell, 1989). Deferoxamine is approved for clinical use as an iron-poisoning antidote under iron-overload conditions, such as a massive transfusion therapy of thalassemia. It is also widely used experimentally as an inhibitor of iron-catalyzed LPO both in vitro and in vivo. Deferoxamine has been shown to penetrate the blood–brain barrier and to accumulate in the brain tissue at a significant concentration quickly after subcutaneous injection (Palmer et al., 1994).
The abundance of superoxide anion radicals, produced during ischemia and reperfusion, can prevent endothelium-dependent dilation of small vessels (arterioles), probably because of inactivation of •NO and •NO regulatory pathways of vascular tone (Hein and Kuo, 1998). This leads to increased vessel reactivity to constricting agents, such as epinephrine, and can at least partially account for microcirculation disorders during postischemic reperfusion. Perivascular application of inhibitors of free radicals, including DFO, led to significant depression of arterial narrowing in a model of femoral artery vasospasm (Luo et al., 1995), suggesting the important contributions of free radical reactions and their products to postischemic vasoconstriction and possibly no-reflow phenomenon. In our study, we discovered that DFO increased cerebral perfusion significantly only during the first 20 minutes after resuscitation. The serum half-life of DFO after a single intravenous injection is very short (5 to 10 minutes) (Summers et al., 1979). This probably explains why the DFO-induced elevation of cerebral blood flow in the present study subsides after 20 minutes. Therefore, the time course of administration of DFO should be carefully considered. Additional injections of DFO 15 minutes after resuscitation or constant infusions of the drug during this period are implicated and deserve further investigation.
We injected DFO 5 minutes before resuscitation, that is, 12 minutes after the beginning of the cardiac arrest. At this point, rats were virtually dead and, presumably, DFO stayed at the injection point (femoral vein and inferior vena cava) without being metabolized and cleared. Later, at the time of ROSC, DFO was distributed throughout the body with the blood flow, and was present at the crucial time point of the initiation of LPO during reperfusion. Several previous studies, which reported no beneficial effect of DFO, implemented different treatment approaches. For example, neurologic damage was not prevented with 50 mg/kg of DFO, injected slowly during a 10- to 15-minute period at least 5 minutes before complete cerebral ischemia in dogs (Fleischer et al., 1987). Similarly, the area of cerebral infarction was not diminished if DFO (50 mg/kg) was administered 0.5 h before and 0.5, 3, and 24 h after hypoxic unilateral carotid occlusion (MacMillan et al., 1993).
Another improvement of the present study over some of the previous studies is that our model of cardiac arrest and resuscitation permits very precise control of the severity and duration of the ischemic event (Liachenko et al., 1998, 2001; Xu et al., 2002), with significantly less variability. The combination of this model with noninvasive measurements of cerebral perfusion during the crucial period immediately after resuscitation allowed us to explore the protective effects of DFO that might have been undetected under circumstances with large experimental variations.
We observed lesser neurologic damage and significantly better survival after longer duration of cardiac arrest in the present study than in other similar studies (Neumar et al., 1995). This is likely attributable to the difference in the method of resuscitation. The method used in this study is capable of reinstating high cerebral perfusion in the early stage of resuscitation (Liachenko et al., 2001). In addition, because the heating of animals was discontinued during the arrest time, the injection of DFO solution or saline, which was at room temperature, led to the decrease of the core temperature of the animals to 34.8 ± 0.4°C before ROSC. Hypothermia, even for a short period, could have contributed to better outcome after cardiac arrest (Colbourne et al., 1999). The use of isoflurane anesthesia up to the point of the induction of CA might also provide some protection against ischemia and reperfusion injury (Blanck et al., 2000).
Histologic damage score was not changed by DFO. Typical ischemic changes to neurons were seen only in the CA1 region of hippocampus, and were not found anywhere else in the brain. Although the CA1 region is known to be selectively vulnerable to ischemic and reperfusion damage, it might not be the crucial area for behavioral neurologic deficit expression. Therefore, it is possible that neurologic disorders in our model are of more functional than structural nature, and DFO was probably able to reverse the former but not the latter. It is also possible that the animals with the highest degree of damage did not survive and were excluded from the NDS assessment.
Footnotes
Acknowledgments:
The authors thank Ms. Martha Zegarra for technical assistance.